Peripheral Primitive Neuroectodermal Tumour of the Maxilla and Paranasal Sinuses: A Rare Clinical Presentation
C.L. Krithika1, S. Sathasivasubramanian2, C. Ponranjini Vedeswari3
1 Senior Lecturer, Department of Oral Medicine & Radiology, SRM Dental College, Bharathi Salai, Ramapuram, Chennai, India.
2 Professor & Head, Department of Oral Medicine & Radiology, Faculty of Dental Sciences, Sri Ramachandra University, Porur, Chennai, India.
3 Assistant Professor, Department of Oral Medicine & Radiology, Thoothukudi Government Medical College, Thoothukudi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. C.L. Krithika, Senior Lecturer, Department of Oral Medicine & Radiology, SRM Dental College, Ramapuram, Chennai-600 089, India.
E-mail: krithika.sekar@gmail.com
Peripheral Primitive Neuroectodermal Tumours (pPNET) are rare aggressive tumours of neural crest cell origin. These tumours are more common in children and young adults. This is a unique report of one such presentation in a 23-year-old lady occurring in the left side posterior maxilla and Paranasal sinuses along with imaging characteristics, histopathologic features, immunohistochemical aspects and management. Though these tumours are rare, they should be considered in the differential diagnosis of rapidly growing soft tissue masses in young adults. Precise diagnosis with timely management is necessary for good prognosis.
Aggressive, Oral cavity, Small round cell tumours
Case Report
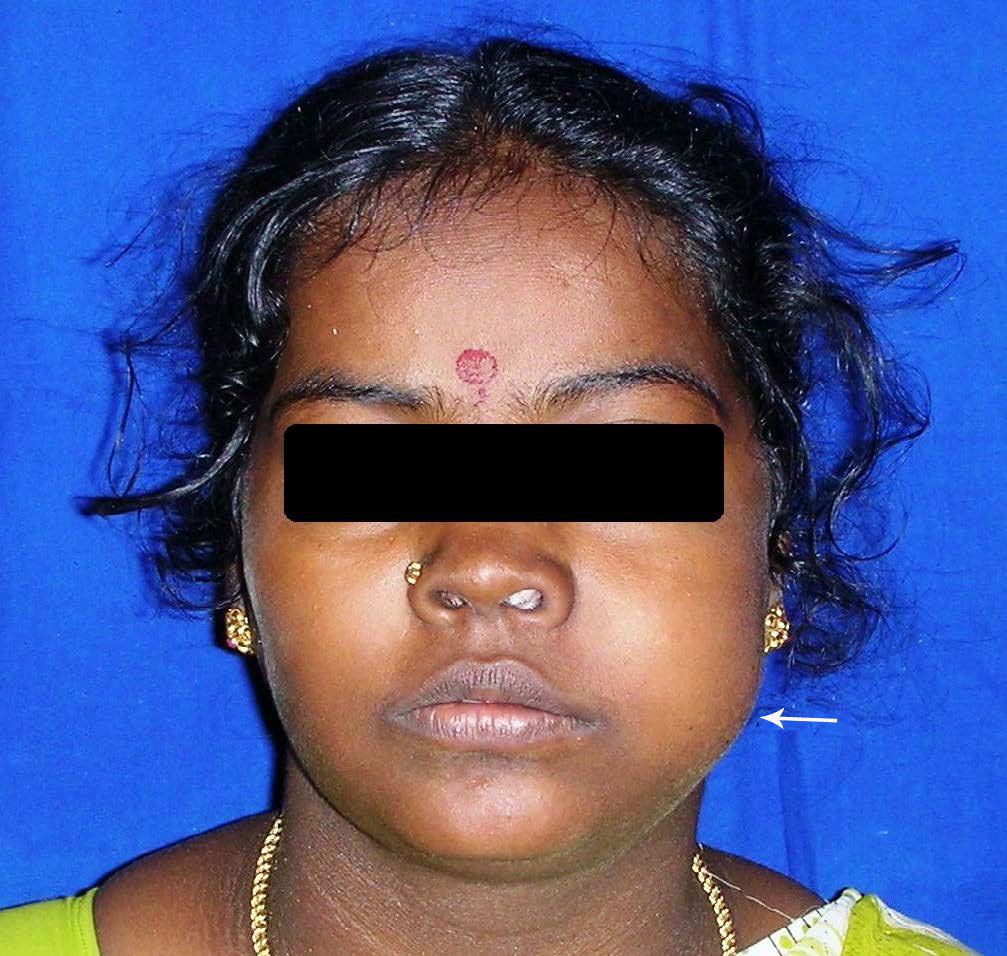
A 23-year-old female patient reported to the Department of Oral Medicine and Radiology, Faculty of Dental Sciences, Sri Ramachandra University, with a complaint of a growth in the left upper back tooth region of 3 months duration. During the last 3 weeks, there was a rapid increase in the size of the growth, with a swelling on the left side of the face. She had associated dull pricking type of pain. After dental consultation she had undergone extraction of two teeth in that region, but did not have any relief of symptoms. She also complained of nasal obstruction and watering of eyes for a duration of two weeks. Medical, family and personal history was not relevant. Extra-oral examination revealed a single diffuse swelling on the left side middle third of the face with obliteration of naso-labial fold [Table/Fig-1]. The swelling was tender and soft to palpate. A single submandibular lymph node was palpable on the left side. It was firm in consistency and movable.
Extra-oral view, with arrow showing the swelling
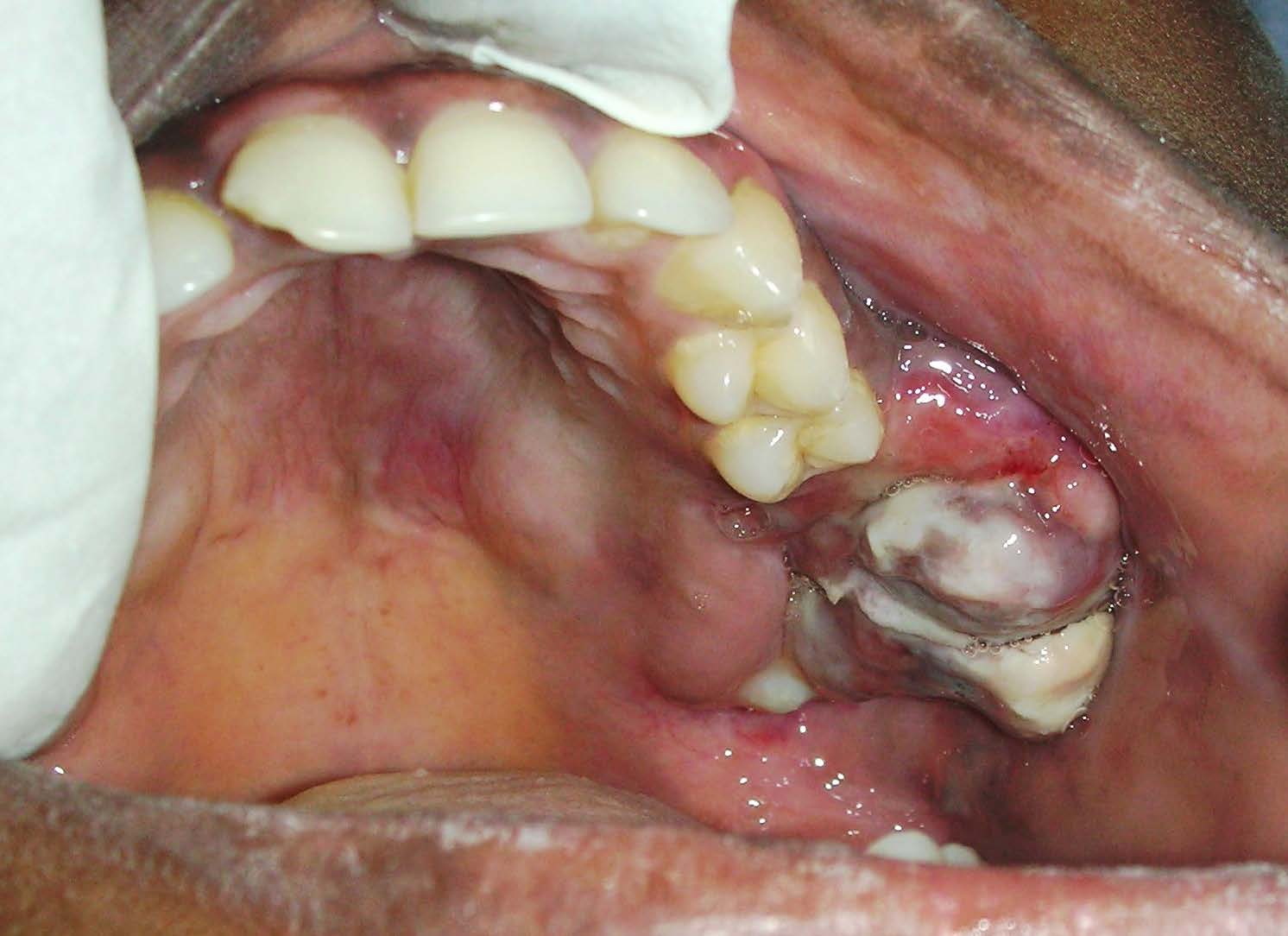
Intraoral examination revealed missing (26&27)and mobile (25&28). A mass was evident in the left postero-lateral aspect of the hard palate in the edentulous ridge region of 26 and 27, roughly measuring about 6 x 3cm in dimension with surface erythema and whitish slough [Table/Fig-2]. It was tender and soft in consistency. Based on the history and clinical presentation, a working diagnosis of a sarcomatous lesion was made and differential diagnosis of minor salivary gland Tumours, and extra-nodal non-Hodgkin’s lymphoma was considered and necessary diagnostic work up was done.
Intraoral view of the lesion
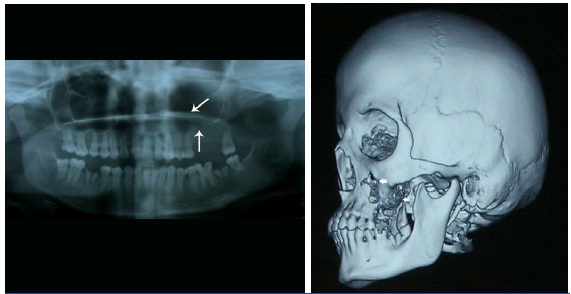
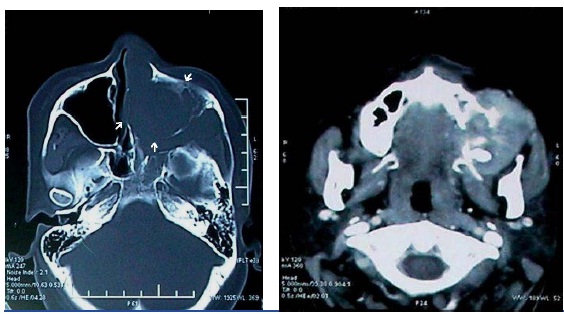
Orthopantomograph revealed opacification of the left maxillary sinus region with destruction of the cortical outline inferiorly and haziness of the left nasal fossa [Table/Fig-3a]. Complete blood count, chest X-ray and other routine investigations for metastatic evaluation were unremarkable. CT revealed a soft tissue density lesion filling the nasal cavity and paranasal sinuses, with bulging of the sinus walls on the left side. There was destruction of anterior and lateral wall of maxillary sinus and alveolar process of maxilla on the left side. Superior, middle and inferior turbinate appeared to be destroyed [Table/Fig-3b&c]. Significant enhancement was seen on contrast administration, with extra-osseous extension of the soft tissue mass [Table/Fig-3d].
Orthopantomograph, with arrows indicating destruction of the cortical outline of the maxillary sinus inferiorly and haziness of the left nasal fossa (b) CT-Three dimensional reconstruction, with arrows revealing destruction of the maxillary alveolus, anterior and lateral wall of left maxillary sinus
CT-axial view – bone window- with arrows revealing a soft tissue density lesion seen eroding and involving the nasal cavity and the maxillary sinus on the left side (d) CT-contrast, axial view- revealing significant enhancement after contrast administration
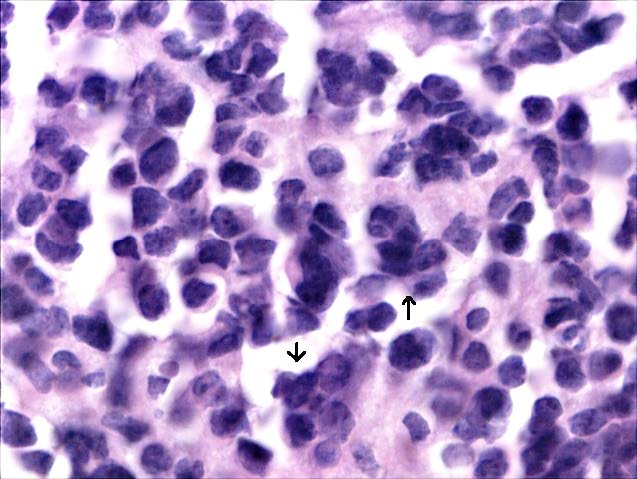
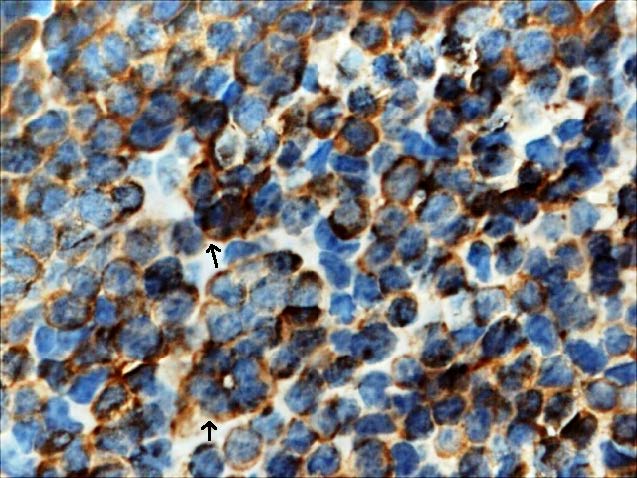
The tumour staging was stage IVA. An incisional biopsy was done and sections revealed monotonous sheets of small blue cells of uniform size, with little monophilic, large nuclei, irregular, hyperchromatic, peppery chromatin and scanty eosinophilic cytoplasm. In some areas, cells were arranged in tiny rosettes at the center with minimal stroma, suggestive of small round cell Tumour [Table/Fig-4a]. Immunohistochemistry revealed positivity for NSE marker [Table/Fig-4b] and was negative for cytokeratins, CD3, desmin, and CD45. Owing to the large size, extension and location of the Tumour, concurrent chemo-radiotherapy was planned rather than surgery. She received chemotherapy with Cisplatin weekly for 6 weeks. Since there was extensive involvement, external beam radiotherapy was also considered. Radiotherapy of 54 Gray was planned. It was administered as one fraction/day, with a dose of 2 Gray per fraction. A total of 4-fractions/ week for 27 sittings were given. Two months after the treatment, a good prognosis was observed. Later the patient was asked to report after 3 months for further metastatic evaluation and management, but lost to follow-up.
H & E section, 100 X view, with arrows revealing the Tumour cells
Tumour cells showing (IHC 100X), with arrows showing Immunopositivity to NSE marker
Discussion
Primitive neuroectodermal Tumours (PNET) are non-epithelia, rapidly growing small round cell malignancies of neural crest cells. Hart & Earle first introduced the term PNET in 1973 [1]. It is classified as peripheral PNET and Central PNET. Central PNETs are tumours that arise from the central nervous system, while peripheral PNETs are derived from tissues outside the central and autonomous nervous system. Peripheral PNETs and Ewing’s sarcoma family of tumours are often interchangeably referred in scientific literature. The microscopic features and genetic changes are the same in Peripheral PNET and extra-skeletal Ewing sarcomas, with chromosomal translocation in t (11:22) (q24:q12). The difference in them is based only on the extent of neuronal differentiation [2]. These tumours can occur at any part of the body, with pelvis being the most common site of occurrence and other sites include chest wall, larynx and abdomen tumours and only 3% of cases have been reported in the skull and jaws [3,4]. They can involve any bone, but predominantly affect central, axial skeleton and diaphysis of long bones. Report of cases involving the jaws are very rare with only 7 reports arising from the mandible [4] in scientific literature and eight cases affecting the maxilla in English literature. These peripheral PNETs constitute to only about 1-4% of soft tissue Tumours [5]. Though there are paucity of cases in the head and neck, the site of presentation is diverse. The clinical symptoms are mainly dependent on the site of presentation, which includes cranial neuropathies, exophthalmos, epistaxis, nasal obstruction and headache. This neoplasm is more common in young adults, especially in the second decade of life with a male predominance [6]. Such lesions are insidious in onset, painful and rapidly progressing leading to large sizes at presentation. These Tumours are highly aggressive and have a high metastatic potential and most of these Tumours show micro-metastasis to bone and bone marrow, making the prognosis very poor.
Clinically, intraoral pPNET have a wide array of differential diagnosis, which includes epulis, pyogenic granuloma, ossifying fibroma, minor salivary gland neoplasms, peripheral giant cell granuloma, carcinoma, non-Hodgkin’s lymphoma, haemangioma and metastatic tumours, but definitive final diagnosis is based on histology and immunohistochemical analysis. Imaging studies are particularly useful in determining the limits of tumour involvement and metastatic evaluation.
On light microscopy, they exhibit narrow sheets of poorly differentiated monotonous collection of small round cells with uniform, oval nuclei and scanty cytoplasm. It is difficult to distinguish pPNETs from other round cell tumours, based on histologic studies alone, immunohistochemical analysis and cytogenetics also play a vital role. The diagnostic criterion includes: (a) Evidence of Flexner- Winter-Steiner/Homer-Wright rosettes, (b) positivity for two or more neural markers and (c) Presence of neuro-secrectory granules and neural differentiation, under electron microscopy [3].
Osteosarcoma can resemble PNET, but they do not exhibit Flexner rosettes and positivity for neuroendocrine markers (NSE, S-100, chromogranin, neurofilaments) and present with characteristic osteoid formation. Lymphomas show positive reaction for CD45, rabdomyosarcomas show positivity for myogenin and desmin. Positive staining for vimentin and CD 99 plays a key role in the differentiation of PNET from other small round cell tumours [3].
The head and neck region with complex anatomic constraints, poses great difficulty in determining the treatment modality with concern for adjacent vital structures and esthetics. Significant prognostic factors include site, Tumour volume and presence of metastasis [7]. The most common sites of metastasis include the lung, bone, and bone marrow. Distant metastasis, especially to bone and bone marrow in 20 to 25% of newly diagnosed cases, has been reported [5]. There is no set protocol for their management; a multimodality approach is necessary, which include chemotherapy and surgery with or without radiotherapy for local and regional control. Chemotherapy along with radiotherapy may be indicated in inoperable cases and tumours that do not respond to neo-adjuvant therapy. Such patients should be followed up for lifetime, to rule out recurrence and metastasis [3]. When treated with chemotherapy, these tumours show intermediate prognosis, as they are highly aggressive and patient may have metastatic disease at the time of presentation. Localized tumours show a 2-3 year-survival rate of 65% to 56% respectively [8]. Outcomes also vary based on disease sub-site and patients with metastatic disease have poor prognosis of 0-25% of 5 year survival rates compared to 40-79% for localized disease [9].
Conclusion
Peripheral PNETs, though being rare in the oral cavity, should be considered in the differential diagnosis of aggressive, soft tissue masses of the jaws, especially in young adults. Early histopathologic diagnosis with timely management plays a crucial role in their prognosis.
[1]. Shi L, Guo Z, Wu X, Primary pulmonary primitive neuroectodermal Tumour metastasis to the pancreas: a rare case with 7-year follow-upDiagnostic Pathology 2013 8:51 [Google Scholar]
[2]. Park IH, Hong SM, Jung JW, Lee HM, A case of primitive Neuroectodermal Tumour in the Nasal CavityJ Rhinol 2013 20(1):62-64. [Google Scholar]
[3]. Monal BY, Tupkari JV, Shubhangi M, Joshi Pradnya, Primitive neuroectodermal Tumour of the posterior mandible: A case reportJournal of Clinical and Experimental Investigations 2013 4(1):101-04. [Google Scholar]
[4]. Arvind K, Vijayalakshmi R, Urmila M, Rinku G, Primary peripheral primitive neuroectodermal Tumour of the mandibleCase Report - Surgical Pathology 2013 3:192-94. [Google Scholar]
[5]. Poornima K, Meenaxi VU, Pascsl XP, Primitive Neuroectodermal Tumour of Maxilla: A rare caseWorld Journal of Dentistry 2011 2:281-83. [Google Scholar]
[6]. Windfuhr JP, Primitive neuroectodermal Tumour of the Head and Neck: incidence, diagnosis and managementAnn Otol Rhinol Laryngol 2004 113:533-43. [Google Scholar]
[7]. Kimber C, Michalski A, Spitz L, Pierro A, Primitive neuroectodermal Tumours: anatomic location, extent of surgery and outcomeJ Paediatric Surg 1998 33:39-41. [Google Scholar]
[8]. Jones JE, McGill T, Peripheral primitive neuroectodermal Tumour of head and neckArch Otolaryngol Head Neck Surg 1995 121:1392-95. [Google Scholar]
[9]. Scurr M, Judson I, How to treat the Ewings family of sarcomas in adult patientsOncologist 2006 11:65-72. [Google Scholar]