Nephropathic Cystinosis Presenting as Renal Fanconi Syndrome without Glycosuria
Jayashree Kanthila1, Smitha Dsa1, Kamalakshi G. Bhat1
1 Associate Professor, Department of Pediatrics, Kasturba Medical College, Mangalore, Manipal University, India.
2 Assistant Professor, Department of Pediatrics, Kasturba Medical College, Mangalore, Manipal University, India.
3 Professor, Department of Pediatrics, Kasturba Medical College, Mangalore, Manipal University, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Kamalakshi G Bhat, Professor, Department of Pediatrics, KMC Hospital, Attavar, Mangalore 575001, India.
E-mail: kamalakshibhat@gmail.com
Renal Fanconi syndrome is diagnosed by its cardinal features of glycosuria without diabetes, aminoaciduria, phosphaturia, and renal tubular acidosis. It is often associated with hypokalaemia, hypophosphatemia and rickets. We report a seven-year-old boy with nephropathic cystinosis who presented with all the cardinal features of renal Fanconi syndrome associated with rickets, pathological fractures, stage IV chronic kidney disease (CKD) and hypothyroidism. Slit-lamp examination of the cornea confirmed the diagnosis. However glycosuria was conspicuously absent. Whenever there are features of rickets with failure to thrive and recurrent vomiting renal rickets should be ruled out. Cystinosis is one such disorder and we report this case due its rarity and interesting clinical presentation.
Cystinosis, Glycosuria, Renal fanconi syndrome
Case Report
A seven-year-old boy, born out of third degree consanguineous parentage with no family history of cystinosis or its components, had presented with isolated motor developmental delay and failure to thrive from six months of age. On examination he had proportionate short stature {Height: 77cm (<3rd centile), Weight: 8.4 kg (expected: 23 kg, <3rd centile), upper segment to lower segment Ratio was 1.08:1}. His head circumference was 47.5 cm [Table/Fig-1].
Short stature in compared with the child of same age

Other findings on examination include frontal bossing, pallor, bilateral clinodactyly, short fourth metatarsals and features of rickets (rickety rosary, widening of wrist joints, pot belly and genu valgum). Laboratory investigations are described in [Table/Fig-2].
Laboratory investigations at initial work up
| Laboratory investigations | Value | Remarks |
|---|
| Haemoglobin | 9.8 gm/dl | |
| Total leucocyte count | 14300 cells/cmm | |
| Platelet count | 324000 cells/cmm | |
| Peripheral smear | Normocytic normochromic anemia | |
| Urine – albumin | +++ | |
| Sugar | Absent | No glycosuria |
| Specific gravity | 1010 | |
| Quanitative – protein | 108mg/m2/hour | proteinuria |
| | |
| Blood urea | 66 mg/dl | |
| Serum Creatinine | 1.6 mg/dl | |
| Electrolytes – sodium | 132meq/dl | |
| Potassium | 2.6 meq/dl | |
| Calcium | 7.8mg/dl | hypocalcaemia |
| Phosphorous | 3.3mg/dl | hypophosphatemia |
| Alkaline phosphatase | 731 u/l | elevated |
| Thyroid function – TSH | 63.2micIU/ml | hypothyroidism |
| Vitamin D | More than 70ng/ml | After receiving vitamin D injection |
| Parathyroid hormone | 108.2pg/ml | |
| Glomerular filtration rate | 26ml/min/1.73m2 | Stage 4 CKD |
| Tubular Reabsorption of Phosphate | 36% | |
| TmP/GFR | 1.48 | |
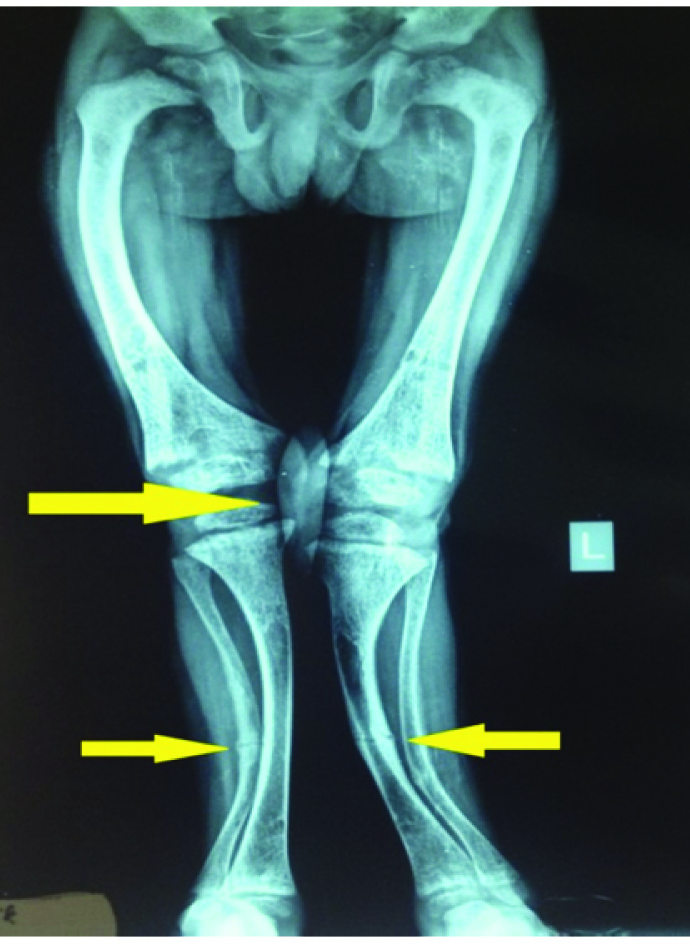
X-Ray of upper and lower limbs had features suggestive of rickets along with pathological fractures involving tibia, fibula and ulna [Table/Fig-3].
X-ray of lower limbs showing features of rickets;
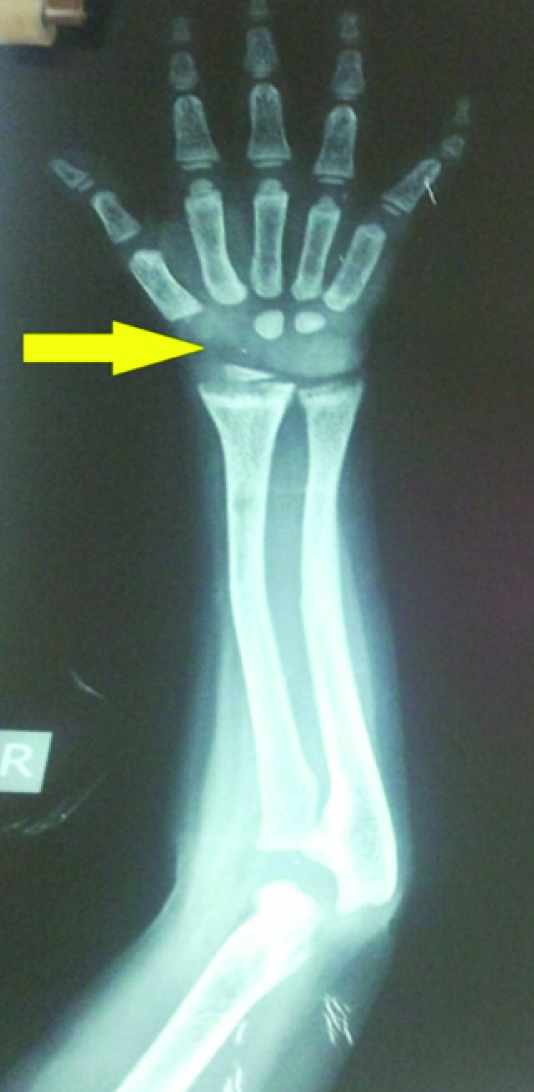
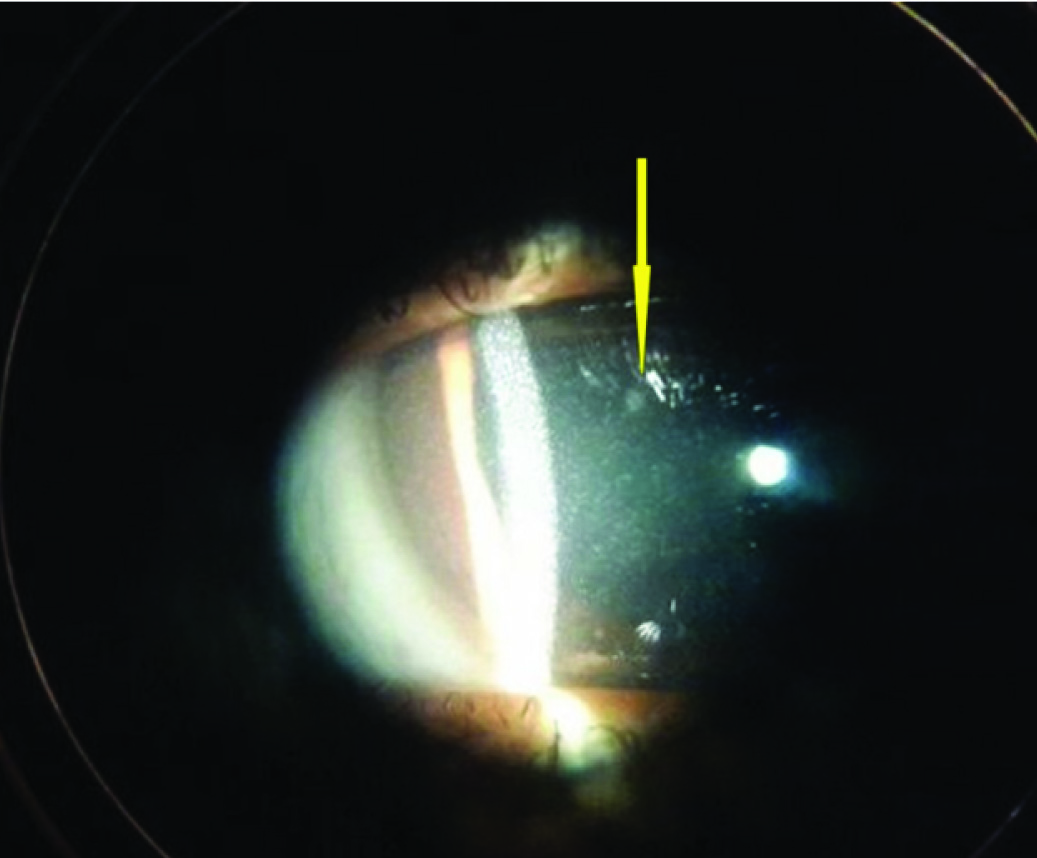
X-Ray B/L wrist had only two carpal bones visualized suggestive of delayed bone age [Table/Fig-4]. USG abdomen and pelvis revealed bilateral grade II renal parenchymal changes with no nephrocalcinosis. Glomerular filtration rate (GFR) was 26 ml/min/1.73 m2 suggestive of stage IV CKD. In the presence of metabolic acidosis, hypophosphatemia, hypocalcemia, hypokalemia, phosphaturia and proteinuria with evidence of hypothyroidism a diagnosis of renal Fanconi Syndrome with hypothyroidism with Stage IV CKD was considered. Ophthalmological evaluation revealed crystal deposits in corneal stroma [Table/Fig-5] and retinal pigment clumping and areas of hypopigmentation in the fundus, which helped us to clinch the diagnosis of Cystinosis. The specific treatment, cysteamine was not available to our patient and hence we could only give supportive care including potassium, calcium, phosphorus supplements along with thyroxin replacement. In the past six months he had two episodes of hypocalcemic tetany associated with febrile illness probably due to inability to take oral supplements. He is on regular monthly follow up and continues to have hypokalemia, hypocalcemia, with no gain in height or weight despite supplements.
X-ray of wrist showing delayed bone age
Corneal cystinosis crystals shown by slit lamp photography
Discussion
Cystinosis is an autosomal recessive disorder characterized by accumulation of amino acid cystine in lysosomes throughout the body, resulting in intracellular crystal formation [1]. The estimated incidence is one in 100,000 to 200,000 live births. Age of onset of symptoms varied from three to 50 y [2]. Depending on the age at presentation and degree of severity, cystinosis is classified clinically into nephropathic infantile form (the most frequent and most severe form), nephropathic juvenile form (intermediate cystinosis, adolescent form) and non-nephropathic adult form (benign non-nephropathic cystinosis, ocular non nephropathic cystinosis).
Clinical symptoms are absent at birth and eventually develop during the initial months of life. The kidneys are the first affected organs manifesting as proximal tubular dysfunction, also known as renal Fanconi syndrome resulting in excessive urinary loss of low-molecular-weight protein, glucose, amino acids, phosphate, calcium, magnesium, sodium, potassium, bicarbonate, carnitine, water, and other small molecules. The diagnosis of cystinosis can be missed during the first months of life as limited number of urinary markers is used to identify renal Fanconi syndrome. However, by six months of age, complete Fanconi syndrome with all cardinal features is usually present.
Cystinosis may present with symptoms of polyuria, thirst, failure to thrive, growth retardation, vomiting, periods of dehydration, constipation and developmental delay. Phosphaturia leads to hypophosphatemic rickets, with characteristic metaphyseal widening, rachitic rosary, frontal bossing, genu valgum, failure to walk, and elevated serum alkaline phosphatase levels. Children with cystinosis may initially receive a diagnosis of Bartter’s syndrome, especially when they present with hypochloremic metabolic alkalosis and hypokalaemia [3,4]. In most patients the GFR remains normal for up to two years and then progressively deteriorates towards end stage renal disease (ESRD) at the end of the first decade [4]. Untreated cystinosis causes systemic complications such as hypothyroidism, (as in our case) one of the first complications to develop in first decade of life and if untreated contributes to growth failure. Photophobia, chronic renal failure, myopathy, difficulty in swallowing, retinal blindness by second to third decade. Diabetes mellitus, male hypogonadism, pulmonary dysfunction, central nervous system calcifications and symptomatic deterioration of central nervous system are the other complications [1].
Cystinosis is diagnosed by determination of elevated cystine levels in PMNs [patients generally have values of 3.0–23.0 nmol half-cystine/mg cell protein (normal <0.2)], eye examination (for corneal crystals) and confirmed by CTNS gene analysis [5]. Patients in developed countries, regardless of their adherence to a drug regimen, generally have expectations of a better quality of life with newly available but expensive long-acting formulations of cysteamine, whereas many patients in developing countries still have poor outcomes because of restricted access to first-generation cysteamine formulations as in our case [6] or may have poor compliance leading to complications [7].
The index case had all the clinical features to suspect nephropathic cystinosis and ophthalmological evaluation confirmed presence of corneal crystals. However, there was no glycosuria, despite having nephrotic range proteinuria with stage IV CKD. Glycosuria with normal blood glucose is one of the cardinal features of renal Fanconi Syndrome. To best of our knowledge nephropathic cystinosis without glycosuria has not been reported hitherto in the literature.
Conclusion
Every case of non-nutritional rickets needs to be evaluated completely before treating with Vitamin D. In children with renal rickets cystinosis needs to be considered as early treatment with cysteamine and appropriate fluid electrolyte management helps in improving the quality of life in these children. Glycosuria, one of the cardinal feature of renal Fanconi syndrome was not present in this case and this could be due to selective tubular dysfunction.
[1]. Gahl WA, Thoene JG, Schneider JA, CystinosisN Engl J Med 2002 (347):111-21. [Google Scholar]
[2]. Servais Aude, Morinière Vincent, Grünfeld Jean-Pierre, Late-Onset Nephropathic Cystinosis: Clinical Presentation, Outcome, and GenotypingClin J Am SocNephrol 2008 (3):27-35. [Google Scholar]
[3]. Pennesi M, Marchetti F, Crovella S, Boaretto F, Travan L, Lazzerini M, A new mutation in two siblings with cystinosis presenting with Bartter syndromePediatr Nephrol 2005 20:217-19. [Google Scholar]
[4]. Ertan Pelin, Evrengul Havva, Ozen Serkan, Emre Sinan, A Patient with Cystinosis Presenting Like Bartter Syndrome and Review of LiteratureIran J Pediatr 2012 22(4):543-46. [Google Scholar]
[5]. Wilmer Martijn J, Schoeber Joost P, Levtchenko Elena N, Van Den Heuvel LP, Cystinosis: practical tools for diagnosis and treatmentPediatr Nephrol 2011 (26):205-15. [Google Scholar]
[6]. Bertholet-Thomas Aurelia, Bacchetta Justine, Nephropathic Cystinosis — A Gap between Developing and Developed NationsN Engl J Med 2014 370:1366-67. [Google Scholar]
[7]. El-Naggari Mohamed A, Elnour Ibtisam, Al-Kindy Hussein, Al-Shahrabally Aamir, Abdelmogheth Anas A, Successful Management of a Neglected Case ofNephropathic CystinosisSultan Qaboos University Med J 2014 14(2):e245-8.Epub. 7TH Apr 14 [Google Scholar]