Perrault Syndrome – A Rare Case Report
Sampathkumar Geethalakshmi1, Narendrakumar Veerasigamani2
1Assistant Professor, Department of General Medicine, Aarupadai Veedu Medical College, Puducherry, Formerly Junior Resident, Government Stanley Medical College Hospital, Chennai, India.
2Formerly Junior Resident, Department of ENT, Madras Medical College and Rajiv Gandhi Government General Hospital, Chennai, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Geethalakshmi S, 1, Jagath Avenue, 6th Main Road Extn, Chromepet. Chennai-600044, India.
E-mail: drgeethalakshmi86@gmail.com
Perrault syndrome is a rare disease comprising pure gonadal dysgenesis (46 XX) and sensorineural hearing loss in females and deafness alone in affected males. It is an autosomal recessive disorder. Over the years many additional features like marfanoid habitus and central nervous system findings have also been reported. Herein we report a case of sporadic Perrault syndrome in 18-year-old female who presented to our hospital with deaf mutism and primary amenorrhoea. On evaluation, the patient had hypergonadotropic hypogonadism, streak gonads and a normal karyotype (46 XX). Audiologic evaluation showed sensorineural deafness. The patient was started on hormone replacement therapy. She is on regular follow up. We present this case for its infrequent incidence and also to add to the ever expanding clinical spectrum of this disease.
Karyotype, Pure gonadal dysgenesis, Sensorineural deafness
Case Report
An 18-year-old female [Table/Fig-1] presented to the outpatient department with history of not attaining menarche. She was deaf mute since birth. She was the eldest child of the family. She was born to parents of consanguineous marriage. Her younger brother aged 16-year-old was 156 cms tall and had normal speech, hearing and age appropriate development of secondary sexual characters. Her mother was 164 cms tall and had attained menarche at 15 y. There was no history of similar complaints or any other significant medical disease in the family.
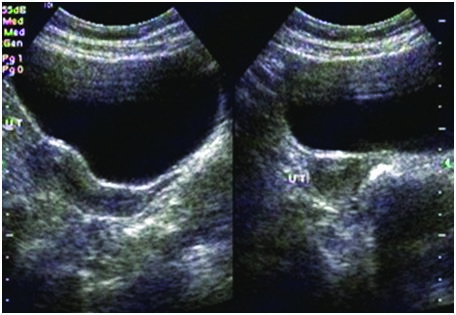
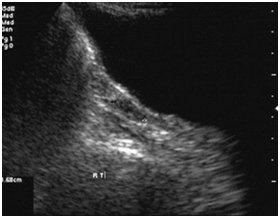
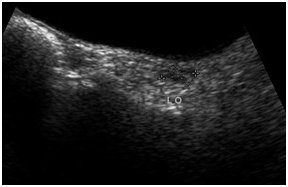
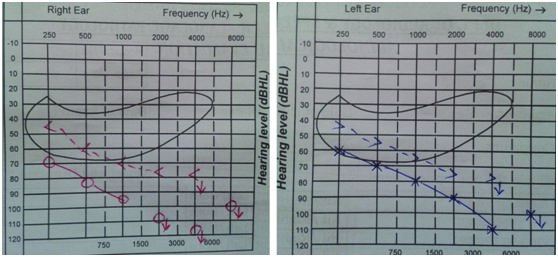
On examination, she had high arched palate, her height was 146 cm, her arm span (153 cm) was more than her height with no other marfanoid features. The patient had poorly developed breasts (Tanner stage 2), sparse pubic and axillary hair (Tanner stage 2). She had normal external genitalia. Her vitals were normal. Systemic examination was normal. Blood investigations showed normal haemogram. Her blood glucose, renal function tests, liver function tests, serum calcium and serum electrolytes were within normal range. Ultrasonogram of abdomen and pelvis showed hypoplastic uterus [Table/Fig-2] and streak ovaries [Table/Fig-3&4]. Echocardiogram revealed normal heart with no evidence of any anomaly. Audiometric evaluation suggested bilateral severe to profound sensorineural hearing loss [Table/Fig-5]. The patient was further subjected to hormonal studies [Table/Fig-6]. She had low levels of estrogen and elevated levels of gonadotropins. Her peripheral blood analysis revealed normal karyotype - 46XX. She was diagnosed with pure gonadal dysgenesis (46 XX) and bilateral sensorineural deafness, thus defining Perrault syndrome. The patient was started on replacement hormonal therapy initially with estrogen and later combined with progesterone. She is being regularly followed up.
Discussion
The association of pure gonadal dysgenesis (46 XX) and sensorineural hearing loss in females was described by Perrault and his associates as early as 1951 [1]. It is a rare genetic disorder. In 1979, Pallister and Opitz reviewed the 14 cases of Perrault syndrome reported till then. They concluded that this disease was autosomal recessive in inheritance [2].
The hallmarks of Perrault syndrome include ovarian dysgenesis in females and sensorineural deafness in both females and males. The karyotype is typically normal in affected individuals. The average age at diagnosis is 22 y following a history of delayed puberty in females associated with deafness [3].
The common pathogenetic relationship between ovarian dysgenesis and sensorineural deafness is not yet defined clearly [4]. Sensorineural deafness is just a part of widespread neurological involvement seen in patients with Perrault syndrome. In a literature review of Perrault syndrome on 21 patients, Nishi et al., reported ataxic gait, pes equinovarus, nystagmus and limited extraocular movements as part of neurological involvement [5]. Recently Stronka et al., reported the association of Leber’s hereditary optic neuropathy with Perrault syndrome [6]. Magnetic resonance imaging in PS patients showed cerebral leucodystrophy, cerebellar hypoplasia, etc. PET scan is helpful in the early detection of the progressive central nervous involvement of perrault syndrome. Fiumara et al., has reported that Perrault syndrome is classified into type I, static, without neurologic features, and type II, with progressive neurologic disease due to its heterogenous nature [7].
The molecular basis of the syndrome is still unclear. The disease may represent a heterogeneous group of genetic disorders with multiple gene defects including HSD17B4, HARS2 gene and CLPP [8]. The genes identified so far were not contiguously located so as to explain the association. Further genetic sequencing and studies are needed to provide a complete panoramic view of this disease. It is important to screen the siblings of the proband so that early identification of the disease might help in timely intervention [9].
Image of the patient showing 18 year female with normal phenotype

Transabdominal ultrasound showing hypoplastic uterus
Rudimentary/streak ovaries on right side
Rudimentary/streak ovaries on left side
Audiogram showing bilateral sensorineural hearing loss
Hormone studies showing elevated gonadotropins
| Name of the Hormone | Patient’s Values | Phase Reference | Range |
|---|
| FOLLICLE STIMULATINGHORMONE(FSH) | 133.61 IU/L | Follicular phase Ovulatory phase Luteal phase | 3.0–20.0 IU/L 9.0–26.0 IU/L 1.0–12.0 IU/L |
| LEUTINISING HORMONE(LH) | 27.16 IU/L Follicular phase | Ovulatory phase Luteal phase | 2.0–15.0 U/L 22.0–105.0 U/L 0.6–19.0 U/L |
| ESTROGEN | 11 pg/ml | Follicular phase Mid-cycle peak Luteal phase | <20–145 pg/mL 112–443 pg/mL <20–241 pg/mL |
| PROGESTERONE | 0.43 ng/ml | Follicular phase Midluteal phase | <1.0 ng/mL 3-20 ng/mL |
| PROLACTIN | 8 ng/ml | | 0–20 ng/mL |
| FREE T3 | | 2.6 pg/ml | 2.4–4.2 pg/mL |
| FREE T4 | | 1.06 ng/ dl | 0.8–1.7 ng/dL |
| THYROID STIMULATING HORMONE(TSH) | | 1.478 mIU/L | 0.34–4.25 mIU/L |
Conclusion
Perrault syndrome may not be rare. Some cases might have been unrecognized or not reported, especially when only one child in a family was affected by this disease. We report this case for its rarity as approximately 40 cases have been reported so far in literature. Most of the cases reported so far have some kind of neurological deficit. However our patient did not have any clinical or radiological neurological abnormalities. The presence of associated marfanoid features has been reported in literature only thrice so far. But our patient didn’t have features other than high arched palate and increased arm span when compared to height. The spectrum of the disease remains incomplete as there are a myriad of features added every day.
[1]. M Perrault, B Klotz, E Housset, Two cases of Turner syndrome with deaf-mutism in two sistersBull Mem Soc Med Hop Paris 1951 67:79-84. [Google Scholar]
[2]. PD Pallister, JM Opitz, The Perrault syndrome: autosomal recessive ovarian dysgenesis with facultative, non-sex-limited sensorineural deafnessAm J Med Genet 1979 4:239 [Google Scholar]
[3]. S Marlin, D Lacombe, L Jonard, Perrault syndrome: Report of four new cases, review and exclusion of candidate genesAm J Med Genet 2008 146A:661-64. [Google Scholar]
[4]. JJ Jacob, TV Paul, SS Mathews, Perrault syndrome with Marfanoid habitus in two siblings.J Pediatr Adolesc Gynecol 2007 20:305-08. [Google Scholar]
[5]. Y Nishi, K Hamamoto, M Kajiyaman, The Perrault syndrome: Clinical report and reviewAm J Med Genet 1988 31:623-29. [Google Scholar]
[6]. M Badura-Stronka, A Wawrocka, K Zawieja, Severe manifestation of Leber's hereditary optic neuropathy due to 11778G>A mtDNA mutation in a female with hypoestrogenism due to Perrault syndromeMitochondrion 2013 13(6):831-34. [A+mtDNA+mutation+in+a+female+with+hypoestrogenism+due+to+Perrault+syndrome+Mitochondrion+2013' target='_blank'>Google Scholar]
[7]. A Fiumara, G Sorge, A Toscano, Perrault syndrome: evidence for progressive nervous system involvementAm J Med Genet A 2004 128A(3):246-49. [Google Scholar]
[8]. EM Jenkinson, AU Rehman, T Walsh, Perrault Syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered proteaseAm J Hum Genet 2013 92(4):605-13. [Google Scholar]
[9]. RA Pagon, MP Adam, HH Ardinger, Perrault syndrome. Gene Reviews. Seattle (WA): University of Washington, Seattle; 1993-2015Available from http://www.ncbi.nlm.nih.gov/books/NBK1116/ (accessed 22Jan 2015) [Google Scholar]