Langerhans cell histiocytosis (LCH) is a disorder associated with proliferation of Langerhans cells in various organs. LCH secondary to multisystem involvement can present in a variety of ways. Because of its infiltrative nature, LCH can involve the skin, lymph nodes, the lung or the liver. Jaundice in LCH is a manifestation of liver disease; biliary dilatation secondary to lithiasis or may be due to coexistent Niemann-Pick disease. However, a case of cholestasis has been very rarely described. Cholestasis may result from lymph nodes obstructing the porta hepatis. In this report, we describe a case of type II histiocytosis X with obstructive cholestasis and pulmonary involvement in the form of cysts without significant lymphadenopathy at the porta.
Case Report
A 2-year-old boy presented with progressive abdominal distention, right ear discharge and seborrhoeic dermatitis since six months of age. Prior to presentation, he also had a one month history of fever, jaundice and rash on his scalp for an uncertain duration. He was appropriate for age; all the anthropometric measurements were within the 50th to 75th centile according to the WHO standards. On admission to the hospital, the patient was febrile, icteric and deeply jaundiced. His urine was positive for bile and stool was acholic. Numerous infiltrative skin lesions were observed on his scalp and back.The lesions were raised, mobile, not attached to underlying structures, and non-tender. Ear examination revealed a dull tympanic membrane and non-foul smelling discharge with a central perforation.
On abdominal examination, prominent veins were noted on the surface. Palpation of abdomen revealed smooth, non-tender, non-pulsatile hepatomegaly which was palpated 3cm below the left costal margin. Firm splenomegaly was noted. Shifting dullness was present. No lymphadenopathy was appreciated on examination of the whole patient.
Investigation
Laboratory findings included mild anemia.Peripheral smear revealed normocytic normochromic RBCs, with no evidence of hemolysis, malarial parasites or abnormal cells. Liver chemistries revealed elevated aspartate transaminase (AST), Alanine Aminotransferase (ALT), alkaline phosphatase (ALP) and Gamma glutamyltransferase (GGT). Total serum, direct fraction and indirect serum bilirubin were increased. Lactate dehydrogenase was 402 IU/l with total cholesterol of 100mg/dl. Renal function tests; including serum electrolytes, urine analysis, Coombs test, and intravenous pyelography were within normal limits. Total serum protein was 6.2gm/dl, serum albumin 3gm/dl and globulin fraction 3.2gm/dl. The INR 1.19 and a Westergen sedimentation rate of 36mm/hr. Bone marrow examination revealed mild plasmacytosis, dyserythropoiesis consistent with hepatic disease but no evidence of metastasis or abnormal cell infiltration [Table/Fig-1]. Cerebrospinal fluid chemistry was normal with no malignant cells appreciated.
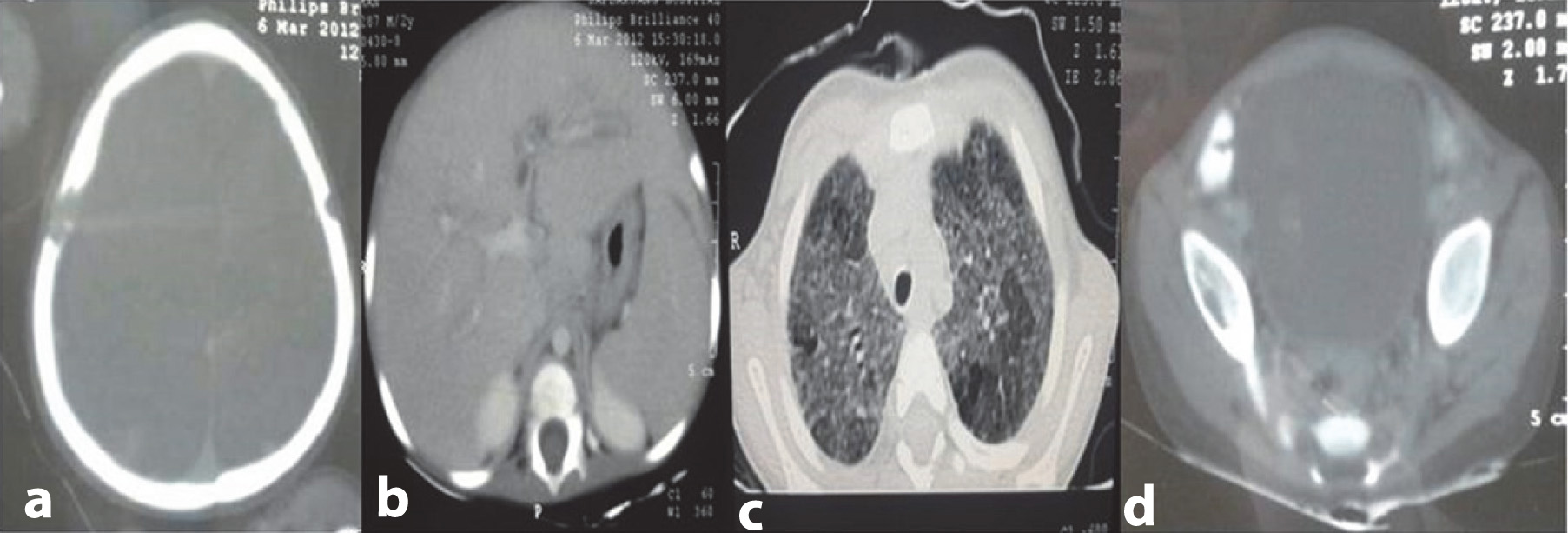
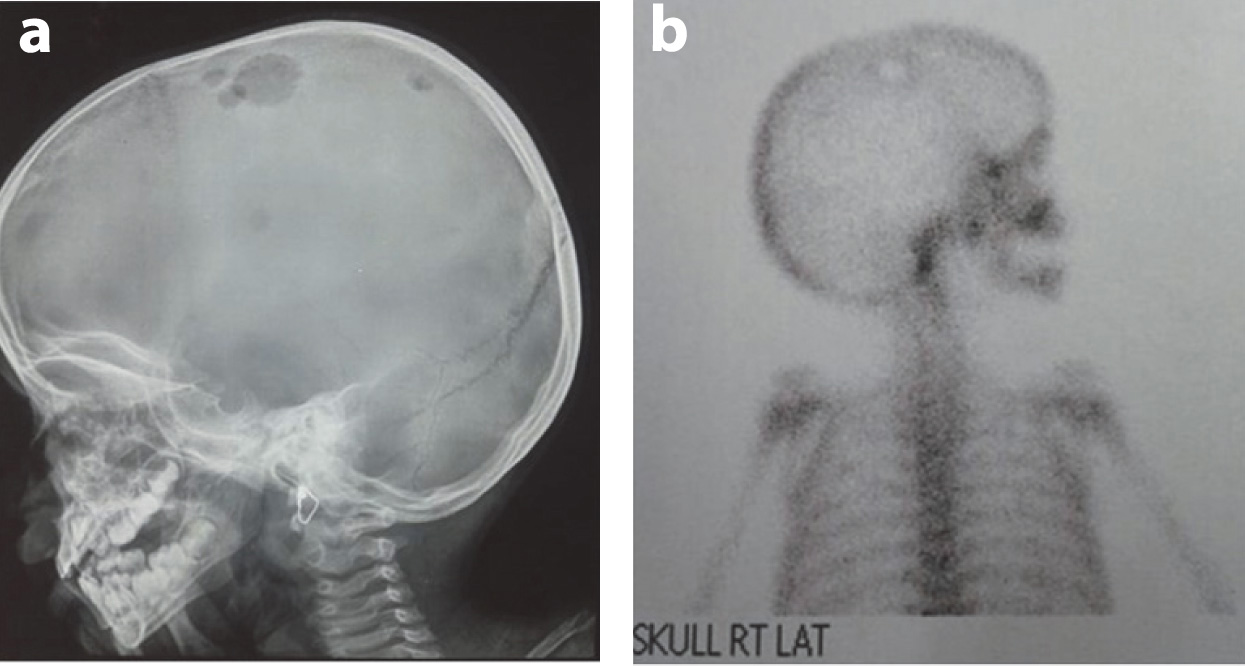
Contrast enhanced tomography of the chest revealed multiple centrilobular nodules in bilateral lung field diffusely exhibiting ground glass opacity with no zonal predominance. However, cystic changes were noted in a few nodules in the upper lobe. Skeletal survey hepatisshowed multiple osteolytic punched out lesions in the parietal areas of the skull bilaterally. A CT of the abdomen revealed moderate hepatosplenomegaly and periportal hypodensities extending along the all portal radicals. Common bile duct, portal vein and gall bladder were visualized as normal. There was no lymphadenopathy or involvement of any other lymph nodes in the area adjacent to the porta hepatis. Lytic lesions in the right iliac crest were prominent [Table/Fig-2a-d]. The 99m TC-MDP bone scan revealed an oval photopenic area surrounded by a rim of mildly increased radiotracer over the right parietal bone and both of the iliacs [Table/Fig-3a,b],
Histology
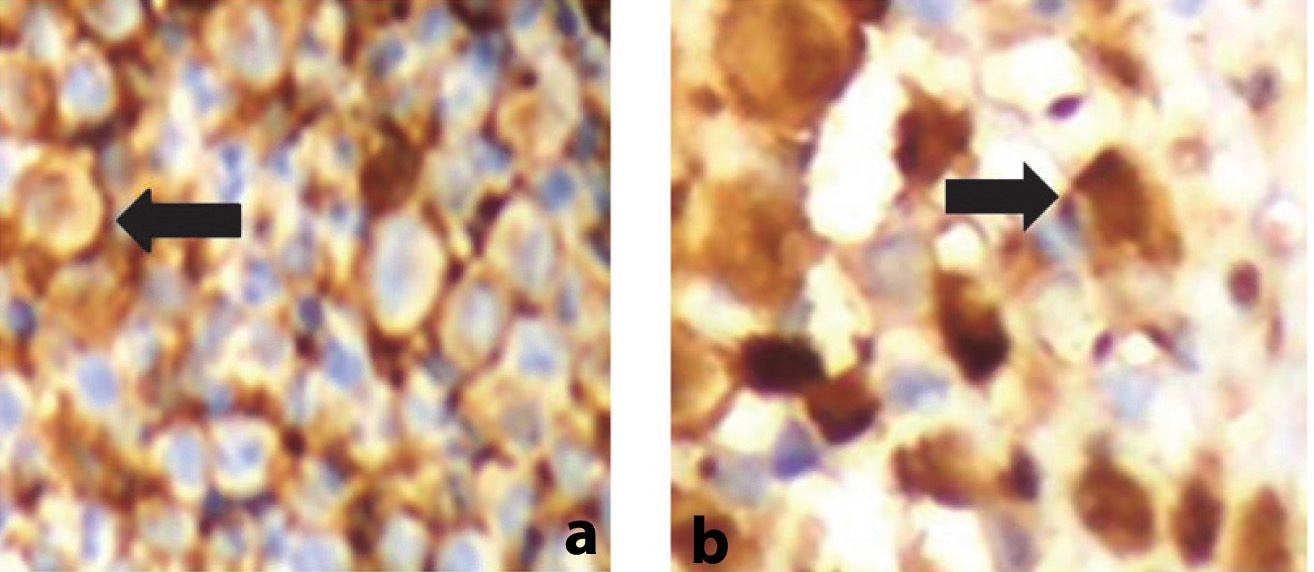
Tissue sample from needle biopsy of the liver revealed periportal infiltration of abnormal cells consistent with histiocytosis X. This was associated with necrosis, multinucleated giant cells, granulomas and eosinophils and involvement of large or medium sized biliary ducts. The skin biopsy of the scalp was read as infiltrate of larger cells with abundant cytoplasm with convoluted to reniform nuclei. Immunohistochemistry showed CD1a positive and S100 positive histiocytic cells [Table/Fig-4a,b].
Therapy was commenced with prednisolone 60mg/m2, vinblastine 6.5mg/m2 and cyclophosphamine 200mg/m2. He completed chemotherapy as per protocol and showed progressive improvement throughout his course and is following in pediatric hemato oncology clinics of the same institute.
Discussion
There are three types of syndromes with histiocyts; LCH, histiocytosis other than LCH and malignant histiocytic disorders [1].
Langerhans cells are antigen presenting cells of the skin and mucous membranes, which have their origin from the bone marrow. These cells are neoplastic and proliferate avidly. They are round cells lacking dendrites, so they are not very apt at antigen presentation [1]. At the focus of inflammation; the release of cytokines by the langerhans cells and other inflammatory cells causes further proliferation and accumulation of more langerhans cells. The released cytokines also stimulate the formation of necrotic foci, osteolysis and the release of connective tissue leading to fibrosis [2].
The median age of presentation of LCH in children has been found to be 30 months [3]. Annually, 2-10 cases per 1 million children are found to suffer from LCH [4,5]. The male and female children are affected in equal proportions [3]. The involvement in LCH can be unifocal, multifocal unisystem or multifocal multisystem and can also involve lungs in the form of pulmonary LCH. Unifocal LCH can be monostotic or polyostotic. Multifocal unisystem variant is associated with fever, bone lesions, diffuse eruptions commonly on the scalp, ear canal and also causes involvement of pituitary stalk causing diabetes insipidus; as is also observed by the authors of previous studies [6]. Multifocal multisystem variant or letterer–siwe disease includes the involvement of bone, endocrine, ocular, central nervous system, spleen, lung and gastrointestinal systems; the last two organ systems were involved in our case scenario. However, more than two-thirds of cases have single system involvement with bone and skin as the most commonly involved sites [5-7].
After a literature analysis in pubmed (Medline); 28 cases of pediatric gastrointestinal LCH have been reported till now, most commonly presenting as hematochezia, followed by non bloody diarrhea, perianal fistula or constipation in that order. Gastrointestinal involvement of histiocytic lesions is rare and includes primary histiocytic disorders of uncertain origin, Rosai-Dorfman disease, LCH, and Erdheim-Chester disease.
The cause of liver pathology in LCH is due to either direct or indirect effects of the langerhans cells. The indirect effects are reversible and occur due to activation of macrophages in our body leading to hepatomegaly, splenomeglay and hypoalbuminemia; however the langerhans cells are absent on liver biopsy [8]. In case of direct liver involvement; there are two types of reported liver pathology on biopsy. The first one is an infiltration of portal tracts without langerhans cells; and the second one is characterized by the domination of langerhans cells in the portal tracts and bile ducts [9] and latter was seen in our case.
On laboratory investigations, the direct involvement category is categorized by cholestasis due to the damage to large or medium sized bile ducts. The disease progression is chronic and leads to the development of sclerosing cholangitis and subsequently biliary cirrhosis. The latter finding was seen in our case.
In a case study of LCH, jaundice has been described rarely. However, LCH can present with earliest features of liver dysfunction; as LCH infiltrates have an affinity for bile ducts causing cholestasis and increased GGT, ALP [10] and bilirubin. Authors of previous studies have reported jaundice in case of LCH in association with hydrops fetalis [11] in a congenital systemic Langerhan cell histiocytosis variant causing excessive hemolysis, hyperbilirubinemia, hypoalbuminemia and failure to thrive in a neonate; as reported by Boccon-Gibod et al., [12]. LCH may also be associated with liver dysfunction with a radiologic appearance of sclerosing cholangitis due to cholangiopathy and stone in the common bile duct causing obstructive jaundice [13]. One case study reports the presence of biliary wall calcification in association with periductal fibrosis and periportal fibrous septa with nodules; consistent with biliary cirrhosis causing jaundice [14]. Another study clues the presence of celiac lymphadenopathy and cholestasis in the setting of LCH; causative of jaundice [15].
There are some conditions which present with jaundice and mimick the presentation of Langerhans cells histiocytosis. These include EBV associated Hemophagocytosis [16], bacterial associated hemophagocytic syndrome in the setting of acute lymphoblastic leukemia [17], non hodgkin lymphoma, acute promyelocytic leukemia and neuroblastoma, respectively [18]. Langerhans Sarcoma like LCH can present as conglomerated lymph nodes due to involvement of the lymph nodes adjacent to the porta hepatis leading to progressive biliary cirrhosis and obstruction; and causing jaundice [19]. This was however not preset in our patient.
Newton and Hamoudi [20] have classified the histiocytosis into two types based on the histological criteria.Type I is associated with widespread infiltrates with individual histiocytes, but not associated with giant cells, eosinophils, or necrosis. In Type II; infiltrates are associated with syncytial like histiocytes and eosinophils as well as foci of necrosis and giant cells. These findings of Type II histiocytosis were appreciated in our case along with interstitial or perivascular cellular infiltrates with fibrosis and cyst formation in the lungs. The tissue biopsy findings were also associated with necrosis, multinucleated giant cells and eosinophils. The definitive diagnosis was determined by staining of lesional cells with CD1a and/or Langerin(CD207) [21-23] rather than the previous demonstration of LC (Birbeck granules) by electron microscopy. Confidence levels for the diagnosis of LCH have been established by the histiocyte society [24].
Most commonly, LCH involves the skin or the lymph nodes; or can involve the lung or the liver. Needle biopsy of our patient’s liver revealed involvement of the periportal areas consistent with type II LCH; as has also been described above.
Pulmonary involvement in our case of histocytosis X was consistent with presence of upper lobe predominant nodules and cysts. The gold standard in the diagnosis of pulmonary histocytosis is surgical lung biopsy [25] Studies have reported that multivisceral involvement is associated with four year survival of 60-80% [24,25].
| Sl. No. | Investigations | |
|---|
| 1 | Hemoglobin | 9.5Gm/dl |
| 2 | Total leucocyte count | 10.7k/ul |
| 3 | Platelet count | 4.5k/ul |
| 4 | Peripheral smear | Normocytic Normochromic Cells,no evidence of hemolysis,malarialparasites,no abnormal cells. |
| 5 | Alkaline aminotransferase (ALT)/Aspartate aminotransferase (AST)/Alkaline phosphatase (ALP)/Gamma glutamyltransferase (GGT)- IU/l | 50/119/4045/204 |
| 6 | S.Bilirubin/Direct fraction/Indirect fraction (gm/dl) | 10/8.9/1.1 |
| 7 | Lactate dehydrogenase | 402U/l |
| 8 | Total serum protein/serum albumin/globulin fraction | 6.2gm/dl/3gm/dl/3.2/dl |
| 9 | INR | 1.19 |
| 10 | ESR (Westergen) | 36mm/hr |
| 11 | Bone marrow examination | Mild plasmacytosis, dyserythropoiesis consistent with hepatic disease, but no evidence of metastasis. | 12 | Cerebrospinal fluid examination (Cytology /Biochemistry gram stain/ Malignant cells) | Normal /Negative for malignant cells. |
(a) Contrast study of head showing lytic lesions involving both of the parietal bones, with bevelled inner edge more pronounced on the right side. (b) Contrast study of abdomen showing moderate hepatoplenomegaly with periportal infiltration at porta hepatis (c) Contrast study of chest showing multiple centrilobular nodules in bilateral lung fields diffusely, producing ground glass opacity; cystic changes appreciated in few nodules in upper lobes; no zonal predominance seen. (d) Lytic lesions in the right iliac crest
(a) Lateral Xray skull showing multiple osteolytic lesion in the parietal bone. (b) Bone scan showing multiple osteolytic lesions in the parietal bone
(a) CD1a positive histiocytic cells. (b) S100 positive histiocytic cells
Conclusion
Although there is multisystem involvement in LCH, Liver involvement in pediatric population is rare; and our case exemplifies the fact that LCH can present as sclerosing cholangitis with features of complete obstructive jaundice without significant lymphadenitis at the porta due to fibrosis and sclerosis of the biliary ducts. Thus health practionners must be cognizant that LCH causing sclerosing cholangitis can present as cholestatic jaundice without significant porta hepatis lymphadenopathy. This knowledge would help to prevent the development of subsequent biliary cirrhosis, portal hypertension and liver failure in their patients; thus ameliorating the need of early liver transplantation.
Acknowledgement
Authors gratefully acknowledge the contribution of Dr. Rakesh Yadav and Dr. KC Aggrawal; Head of the Department of the institution in the study.
[1]. M Sabib, S Ettair, N Erreimi, N Mouane, Sclerosing cholangitis revealing Langerhans cell histiocytosis in a 15-month-old childArch Pediatr 2011 18(9):974-78. [Google Scholar]
[2]. C Bihari, A Rastogi, A Rajbongshi, V Sood, R Khanna, S Alam, Langerhans cell histiocytosis (LCH) presenting as decompensated liver disease: case reportJournal of GHR. 2013 212(8):479-48. [Google Scholar]
[3]. A multicentre retrospective survey of Langerhans’ cell histiocytosis: 348 cases observed between 1983 and 1993. The French Langerhans’ Cell Histiocytosis Study Group Arch Dis Child 1996 75(1):17-24. [Google Scholar]
[4]. H Carstensen, K Ornvold, The epidemiology of Langerhans cell histiocytosis in children in Denmark, 1975-89.Med Pediatr Oncol 1993 21(5):387-88. [Google Scholar]
[5]. JA Salotti, V SaloNanduritti, MS Pearce, Incidence and clinical features of Langerhans cell histiocytosis in the UK and IrelandArch Dis Child. 2009 94(5):376-80. [Google Scholar]
[6]. K Windebank, V Nanduri, Langerhans cell histiocytosisArch Dis Child 2009 94:904-08. [Google Scholar]
[7]. A Guyot-Goubin, J Donadieu, M Barkaoui, S Bellec, C Thomas, J Clavel, Descriptive epidemiology of childhood langerhans cell histiocytosis in france, 2000-2004Pediatr Blood Cancer 2008 51:71-75. [Google Scholar]
[8]. T Gey, C Bergoin, N Just, T Paupard, D Cazals-Hatem, KH Xuan, Langerhans cell histiocytosis and sclerosing cholangitis in adultsRev Mal Respir 2004 21:997-1000. [Google Scholar]
[9]. SL Guthery, JE Heubi, Liver involvement in childhood histiocytic syndromesCurr Opin Gastroenterol 2001 17:474-78. [Google Scholar]
[10]. DG Liu, YX Zhang, F Sheridan, Multisystem Langerhans cell histiocytosis with liver dysfunction as the first presentation: A case reportOncol Lett 2012 3(2):391-94. [Google Scholar]
[11]. CH Lee, TK Lau, KF To, HS Lam, AW Chan, PC Ng, Congenital systemic Langerhans cell histiocytosis presenting as hydropsfetalisActa Paediatr 2005 94(12):1843-47. [Google Scholar]
[12]. LA Boccon-Gibod, HA Krichen, LM Carlier-Mercier, JF Fontaine, JL Leverger, Digestive tract involvement with exudative enteropathy in Langerhanscell histiocytosisPediatrPathol 1992 12(4):515-24. [Google Scholar]
[13]. S Caruso, R Miraglia, M Spada, A Luca, B Gridelli, Biliary dilatation secondary to lithiasis in a child affected by Langerhans’ cell histiocytosis J Clin Ultrasound 2009 37(6):366-68. [Google Scholar]
[14]. S Caruso, R Miraglia, L Maruzzelli, A Luca, B Gridelli, Biliary wall calcification in langerhans cell histiocytosis: report of two casesPediatr Radiol 2008 38(7):791-94. [Google Scholar]
[15]. SW Choi, BS Bangaru, CD Wu, JL Finlay, Gastrointestinalinvolvement in disseminated Langerhans cell histiocytosis (LCH) with durable complete response to 2-chlorodeoxyadenosineand high-dose cytarabineJ Pediatr Hematol Oncol 2003 25:503-06. [Google Scholar]
[16]. JS Chen, CC Tzeng, CJ Tsao, WC Chen, TY Jung, IJ Su, Clonal karyotypeabnormalities in EBV-associated hemophagocytic syndrome Haematologica 1997 82(5):572-76. [Google Scholar]
[17]. KJ Kaplan, ZD Goodman, KG Ishak, Liver involvement in Langerhans’ cell histiocytosis: a study of nine cases Mod Pathol 1999 12(4):370-78. [Google Scholar]
[18]. BW Chen, MH Chang, DT Lin, KH Lin, WM Chuu, KS Lin, Extrahepatic biliaryobstruction caused by cancer of non-liver origin in children: report of 5 casesTaiwan Yi Xue Hui ZaZhi 1989 88(8):819-23. [Google Scholar]
[19]. DC Woong, AI Soo, GC Nak, SP Gyeong, Langerhans cell sarcoma in two young children: Imaging findings on initial presentation and recurrenceKorean J Radiol 2013 14(3):520-24. [Google Scholar]
[20]. W A Newton, A.B. Hamoudi, Perspect, Paediat. Path 1973 :251 [Google Scholar]
[21]. K Chikwava, R Jaffe, Langerin (CD207) staining in normal pediatric tissues, reactive lymph nodes, and childhood histiocytic disordersPediatrDevPathol 2004 7(6):607-14. [Google Scholar]
[22]. SK Lau, PG Chu, LM Weiss, Immunohistochemical expression of LangerininLangerhans cell histiocytosis and non-Langerhans cell histiocytic disordersAm J SurgPathol 2008 32(4):615-19. [Google Scholar]
[23]. R Jaffe, LM Weiss, F Facchetti, Tumors derived from Langerhans cells. In: Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. editorsWHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Lyon: IARC Press 2008 :358-60. [Google Scholar]
[24]. Langerhans Cell Histiocytosis, Histiocyte Society Evaluation and Treatment Guidelines April 2009 Contributors: MilenMinkov Vienna, Austria [Google Scholar]
[25]. XL Ma, KL Shen, B Wang, A child with pulmonary and liver Langerhans’-cell histiocytosisChin Med J (Engl) 2012 125(9):1675-6. [Google Scholar]