Robertsonian Translocation t (21; 21) in a Female Born to Normal Parents: A Case Report
Giriraj Kusre1, Mukul Sarma2, Tulika Nirmolia3, Priyanka Shankarishan4
1Associate Professor, Department of Anatomy, Assam Medical College, Dibrugarh, India.
2Associate Professor, Department of Anatomy, Assam Medical College, Dibrugarh, India.
3Senior Research Fellow, Department of Anatomy, Assam Medical College, Dibrugarh, India.
4Scientist-B, Department of Anatomy, Assam Medical College, Dibrugarh, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Giriraj Kusre, Associate Professor, Department of Anatomy, Assam Medical College, Dibrugarh-786002, India.
E-mail: giriraj_kusre@rediffmail.com
Downs syndrome (DS) occurs due to an extra copy of chromosome 21. About 3% of cases of Downs syndrome occur due to Robertsonian translocation, most commonly t (14; 21), other types of translocations are very rare cause of the syndrome. A 10-year-old patient with mental retardation was admitted following road traffic accident. Patient had flabby muscles, had delayed mile stones, stunted growth for the age, slanting of eyes, flat nasal bridge, and ineligible speech. On cytogenetic analysis the patient had karyotype showing one normal chromosome 21 and one Robertsonian translocation t (21; 21). Parents and siblings of the patient were phenotypically normal. Robertsonian translocation t (21; 21), can occur by transmission from carrier parent, due to ovarian mosaicism for Robertsonian translocation or may appear de novo. In the present case as the parents had normal karyotype and siblings were phenotypically normal, Robertsonian translocation probably have arisen de novo. The present case was a case of Downs syndrome with Robertsonian translocation t (21;21) probably arising de novo.
De novo, Downs syndrome, Mental retardation
Case Report
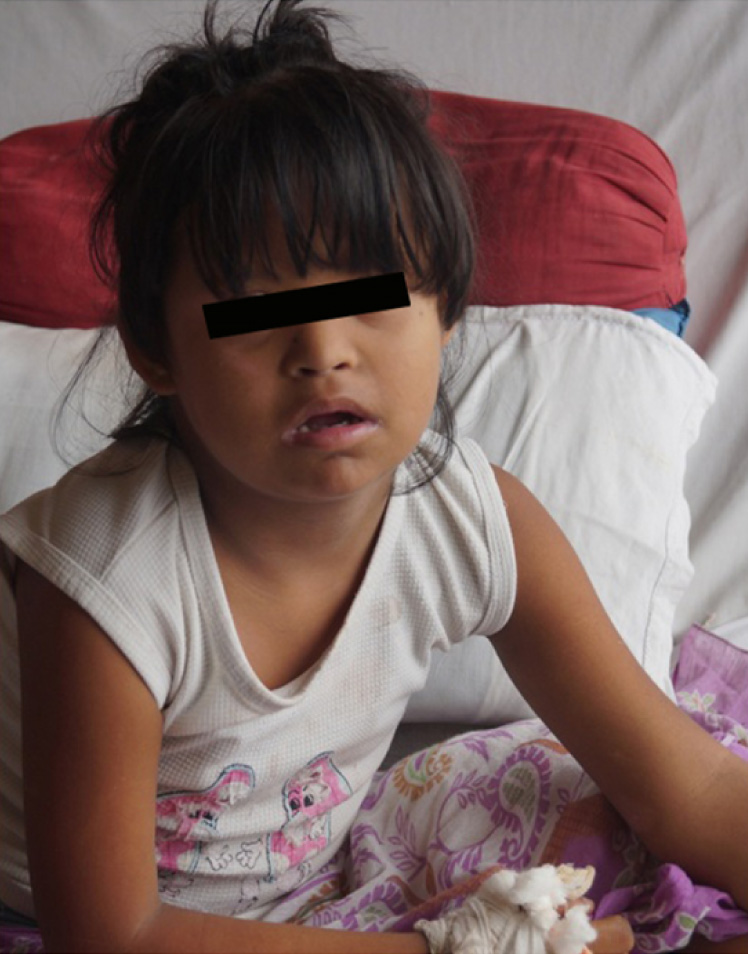
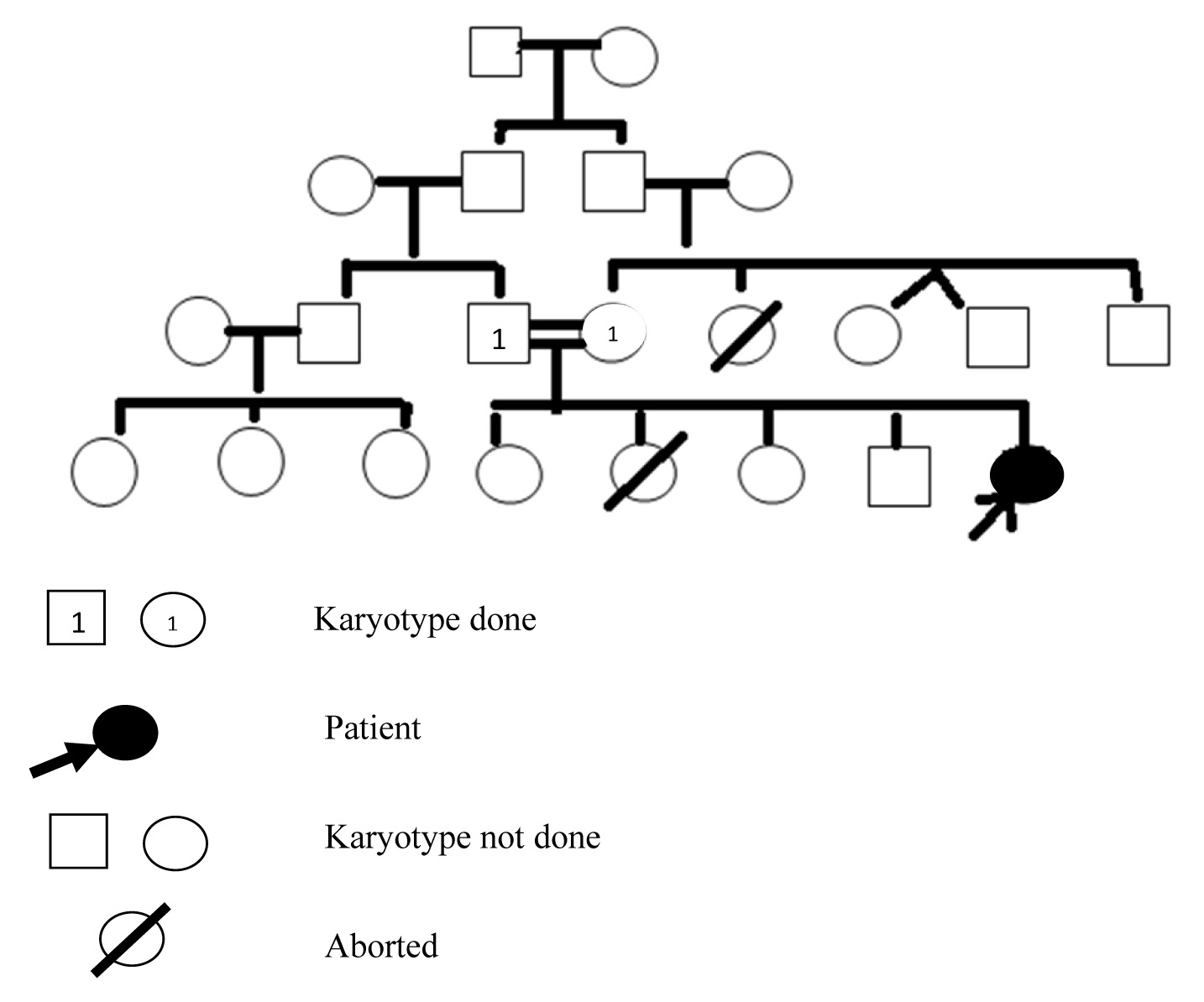
A 10-year-old female patient was admitted in the hospital with a history of trochanteric fracture of femur after road traffic accident. On examination patient was observed to have mental retardation and had Ventricular Septal Defect in the heart. The patient was born with flabby muscles, had delayed mile stones, stunted growth for the age, slanting of eyes, flat face and nasal bridge, ineligible speech, and had difficulty in carrying out her day-to-day activities [Table/Fig-1]. The case was referred to Diagnostic Genetic Lab, Assam Medical College, Dibrugarh for cytogenetic evaluation. The patient was the fourth living child of the consanguineous parents. Other three siblings of the case were normal and were pursuing their studies normally [Table/Fig-2]. Parents of the case were third degree consanguineous. No other family members had any history of mental retardation. The history of the pregnancy during the present case was normal. The results of all biochemical tests were found to be normal.
After obtaining signed informed consent, 3 ml of whole blood was collected in heparinised tube from both parents for lymphocyte culture and karyotype was prepared. The blood was cultured in RPMI 1640 with PHA medium (Peripheral Blood Karyotyping Medium, with Phytohemagglutinin (PHA-M), CAT No # 01-201-1B, Biological industries,) and harvested after 67 and half hours. Slides were prepared for GTG banding. The slides were stained by Giemsa stain (CAT No # S011-100ML, Himedia) and 20 spreads for each of the blood sample were examined for chromosomal abnormality by cytovision workstation (Leica DM6000B, Leica Microsystems).
On cytogenetic evaluation the patient was diagnosed as a case of Downs syndrome with Robertsonian translocation (21; 21) (q10:q10) [Table/Fig-3] whereas the karyotypes from both the parents were normal.
Discussion
About 3.7% of the DS cases occur due to robertsonian translocation with t (14; 21) being the most common one [1]. Robertsonian are the whole-arm exchanges between the short arms of the acrocentric chromosomes (human chromosomes 13–15, 21 and 22) [2]. Although all acrocentric chromosomes may participate in Robertsonian, the distribution of the different possible translocations in the population is non-random [3]. Specifically, rob (13q14q) and rob (14q21q) are the most common, constituting ~ 85% of all Robertsonian [3]. As the present case had Robertsonian t (21;21) (q10:q10), it was a rare type of presentation of downs syndrome.
DS due to translocation may either be de novo or inherited from a balanced carrier parent [4]. A carrier with t (21q;21q) balanced translocation will always have a Downs offspring [5]. In general, carriers of Robertsonian translocations are phenotypically normal. In ~50% of cases of Robertsonian, the rearrangements occur de novo [6] and ~95% of the de novo cases originate during maternal meiosis. Mark et al., [7] reported a case of phenotypically normal woman who had recurrence of DS in her offspring. On cytogenetic analysis of this woman she had normal karyotype from her peripheral blood lymphocyte but had t (21;21) mosaicism (with a frequency of 10% cells) in her ovarian and skin cells. Hulten et al., [8] observed that most females might be having low grade trisomic 21 ovarian mosaics with an average of 0.54% trisomy 21 cells and stated that this ovarian mosaicism may predispose to trisomy 21 conception. In the present case peripheral blood lymphocytes from both parents were analysed. Consent for cytogenetic analysis of other children in the family could not be obtained. Cytogenetic analysis from maternal ovarian cells were technically not feasible, hence were not analysed. As in the present case, the peripheral blood lymphocytes from both the parents had normal karyotype, the trisomy 21 due to Robertsonian translocation (21:21) must have arisen either de novo or due to ovarian mosaicism. As all other siblings of the patient were phenotypically normal, the Robertsonian translocation (21; 21) most probably have arisen de novo.
Photograph of the patient, showing the down’s syndrome features
Pedigree chart of the patient
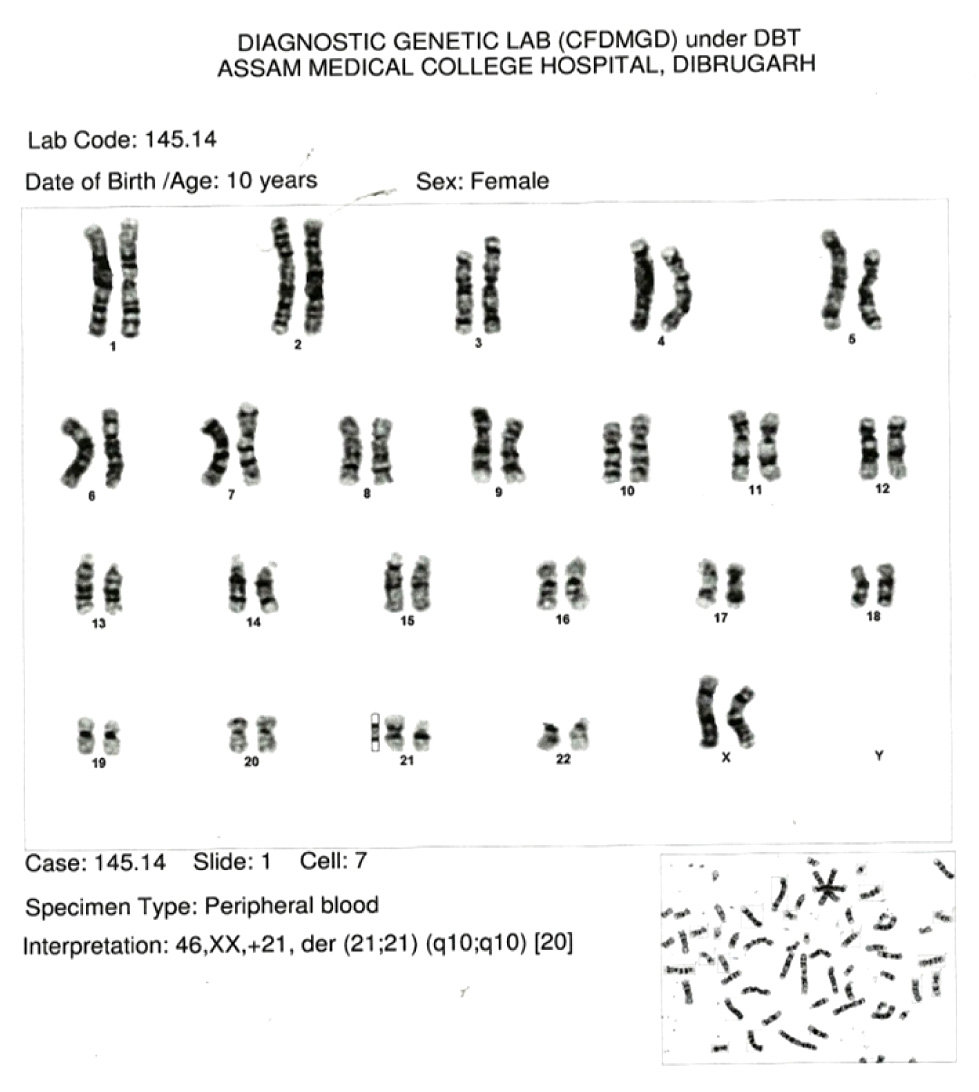
Conclusion
The present case was a case of Downs syndrome occurring due to Robertsonian translocation (21; 21). Both the parents of the patient were phenotypically and cytogenetically normal. Siblings of the case were phenotypically normal. So the present case of Downs syndrome occurring due to Robertsonian translocation (21; 21) probably have arisen de novo.
Acknowledgment
We acknowledge the Department of Biotechnology, Government of India for providing fund to establish the cytogenetic lab where the cytogenetic analysis was done.
[1]. D Mutton, E Alberman, EB Hook, The National Down Syndrome Cytogenetic Register and The Association of Clinical Cytogeneticists: Cytogenetic and epidemiological findings in Down syndrome, England and Wales 1989 to 1993J Med Genet 1996 33:387-94. [Google Scholar]
[2]. R Bandyopadhyay, A Heller, C Knox-DuBois, C McCaskill, SA Berend, L Page Scott, Parental origin and timing of de novo robertsonian translocation formationAm J Hum Genet 2002 71:1456-62. [Google Scholar]
[3]. E Therman, B Susman, C Denniston, The nonrandom participation of human acrocentric chromosomes in Robertsonian translocationsAnn Hum Genet 1989 53(Pt 1):49-65. [Google Scholar]
[4]. J Grouchy, C Turleau, Autosomal disorders. In: Emery AEH, Remoin DL, eds. Principles and practice of medical genetics 1990 New YorkChurchill Livingstone:24771 [Google Scholar]
[5]. AH Emery, RF Mueller, Elements of medical genetics. Student’s NoteGenetics and the physician 1992 8th EditionEdinburghChurchill Livingstone:279-321. [Google Scholar]
[6]. LG Shaffer, CK Jackson-Cook, BA Stasiowski, JE Spence, JA Brown, Parental origin determination in thirty denovo Robertsonian translocationsAm J Med Genet 1992 43:957-63. [Google Scholar]
[7]. HFL Mark, T Mendoza, D Abuelo, LJ Beauregard, JB May, PH Lamarche, Reproduction in a woman with low percentage t(21q21q) mosaicismJ of Med Gen 1977 14(3):221-23. [Google Scholar]
[8]. MA Hulten, SD Patel, M Westgren, N Papadogiannakis, AM Jonsson, J Jonasson, On the paternal origin of trisomy 21 Down syndrome Mol Cytogenet 2010 3:4 [Google Scholar]