Optimizing the Roche LightCycler(R) for Single-Tube Multiplexed RT-PCR Assays
Clarise R. Starr1, Elia T. Villazana2, Richard R. Chapleau3, David L. Masserang4
1 Applied Technology & Genomics Center, United States Air Force School of Aerospace Medicine, 711th Human Performance Wing, Air Force Research Laboratory, Wright-Patterson AFB OH.
2 Applied Technology & Genomics Center, United States Air Force School of Aerospace Medicine, 711th Human Performance Wing, Air Force Research Laboratory, Wright-Patterson AFB OH.
3 Applied Technology & Genomics Center, United States Air Force School of Aerospace Medicine, 711th Human Performance Wing, Air Force Research Laboratory, Wright-Patterson AFB OH.
4 Applied Technology & Genomics Center, United States Air Force School of Aerospace Medicine, 711th Human Performance Wing, Air Force Research Laboratory, Wright-Patterson AFB OH.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Clarise R. Starr, 2510 Fifth Street, Bldg 840, Area-B, Wright-Patterson AFB OH-45433.
E-mail: clarise.starr@us.af.mil
Duplex molecular assay, Duplex PCR, LightCycler, Real-time RT-PCR
Introduction
The Roche Diagnostics LightCycler® (LC) Carousel-Based Systems are a single-source thermocycler capable of detecting light emissions from fluorescing molecules released during RT-PCR. This technology utilizes a single light source at 470 nm to excite fluorophores for read-out in one of three channels (530, 640 and 705 nm) for older 1.5 models and up to six channels (530, 560, 610, 640, 670, and 705nm) for the 2.0 model. The persistent challenge of LC technology is multiplexing, the ability to discriminate more than one target with different fluorophores in a single capillary tube that can be excited at the same 470-nm wavelength and read on 3 or 6 different channels without cross-talk or “bleed-over”. The most commonly employed fluorescent probe is 6-carboxyfluorescein (FAM), fluorescing in channel 1 (530 nm), with bleed-over into the other channels, which is currently compensated for by surrendering valuable carousel space for extraction controls, amplification controls, and duplicate sample runs to assure the generated answer is correct.
Using the 705 nm channel requires resonance transfer probes, as few fluorescent molecules exist with Stokes’ shifts wide enough to achieve detection in this channel. In 2009, a fluorescence resonance energy transfer (FRET)-based method was developed which, when combined with TaqMan style probes, provides a linker that emits a signal that can be read in the unused third channel of the LC 1.5 [1]. This “FRET-MAN” technology works well; however, designing assays with dual probes and quenchers can be challenging. To address this challenge, Biosearch Technologies designed a probe called the Pulsar® 650, which is a fluorescent reporter dye based on a Ru (bpy)3 complex with a large Stokes shift, (λex = 460 nm, λem = 650 nm) that can be used in place of a two-dye FRET construct. Importantly, this dye is stable under oligonucleotidesynthesis conditions, strong acids, and strong bases (Biosearch Technologies, http://www.biosearchtech.com).
We present here an extension of a duplexed assay originally developed for the LC 1.2 [2] to an RNA-based viral target with the inclusion of an internal control for robust quantitation. Here, the primary target was one of two strains of influenza A, chosen due to the increase of awareness of public health in recent years. The third-channel duplex probe was designed based on the human RNase P (hRP), GAPDH, 18S ribosomal RNA, β-actin, cyclophilin or gyrase [3–8]. To demonstrate the ability of duplexed detection, inclusion of the internal controls (ICs) is shown herein to function in both laboratory buffered samples and in clinical matrices. Theoretically, the second probe could instead be used as a detector for a second target organism.
Materials and Methods
Influenza A H1N1 and H3N2 were purchased from American Type Culture Collection (Manassas, VA). Human nasal wash from healthy individuals were obtained from Bioreclamation, Inc. (Westbury, NY) and used as both clinical matrices and positive ICs. Additional flu positive nasal wash samples were obtained from a repository housed by the US Air Force School of Aerospace Medicine Epidemiology program. Viruses were diluted to known concentrations in PBS or spiked into clinical samples and extracted using the BiofireQFLOW” DNA or RNA Sample Purification Kit (Salt Lake City, UT), or by using the automated Qiagen BioRobot M48 Workstation (Valencia, CA). Primers and probes were identified from the literature [4–8], purchased from Biosearch Technologies (Novato, CA) and used at 20 μM and 5 μM, respectively. Assays were run in duplicate on Roche Light Cycler® 1.5.
Results and Discussion
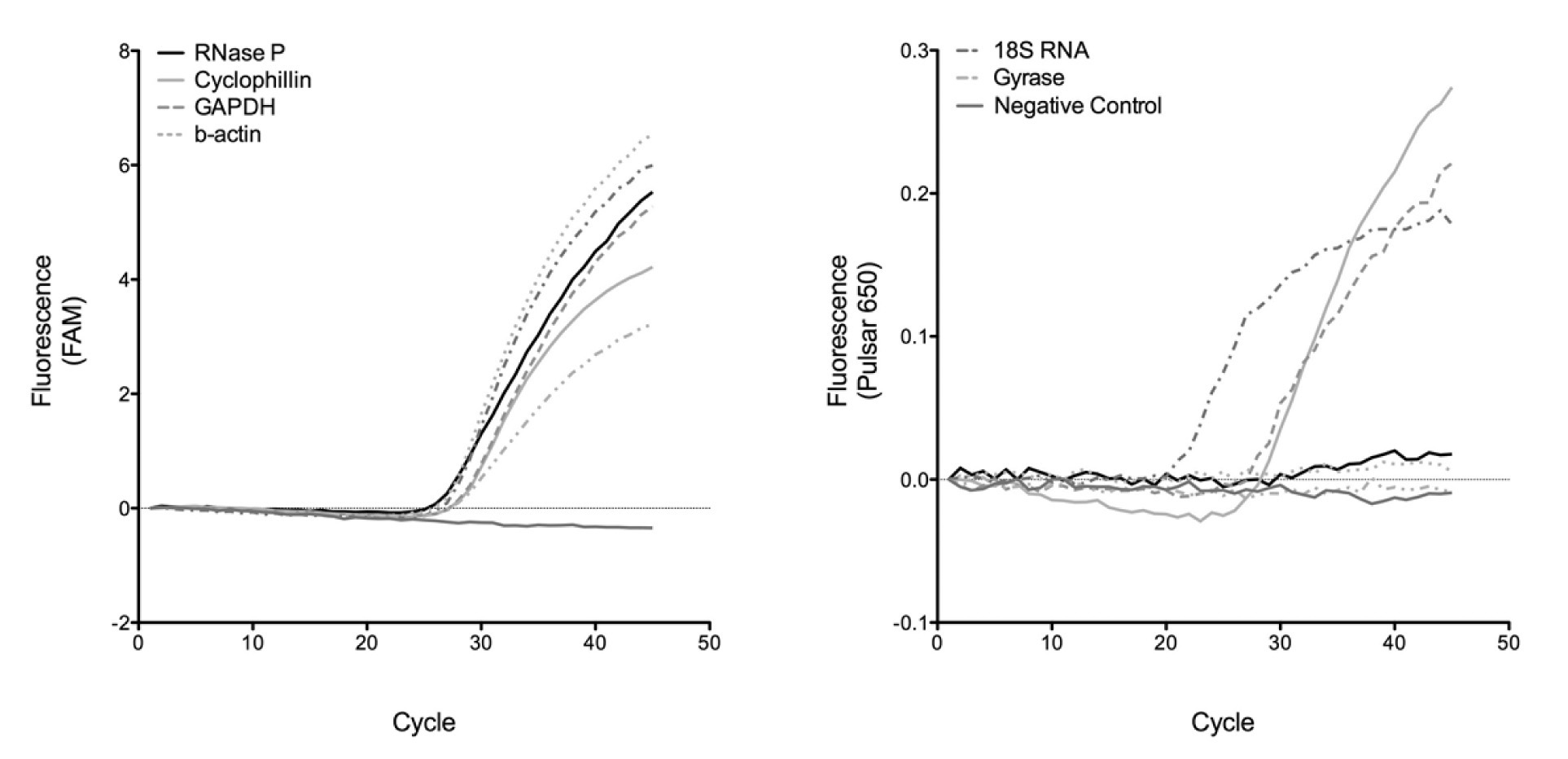
Several IC candidates were evaluated for their efficacy in demonstrating duplex PCR assays on the LC1.5 with varying degrees of success. Gyrase never successfully amplified in any duplex assay and the β-actin IC worked only weakly [Table/Fig-1]. In comparison, duplex assays containing influenza A together with sequences for 18S RNA, GAPDH, hRP and cyclophilin were capable of strongly detecting both signals [Table/Fig-1]. The success of these four ICs demonstrates that using the third channel of the LCis a robust method to screen for successful and inactive duplex assays, due to no cross-channel bleed.
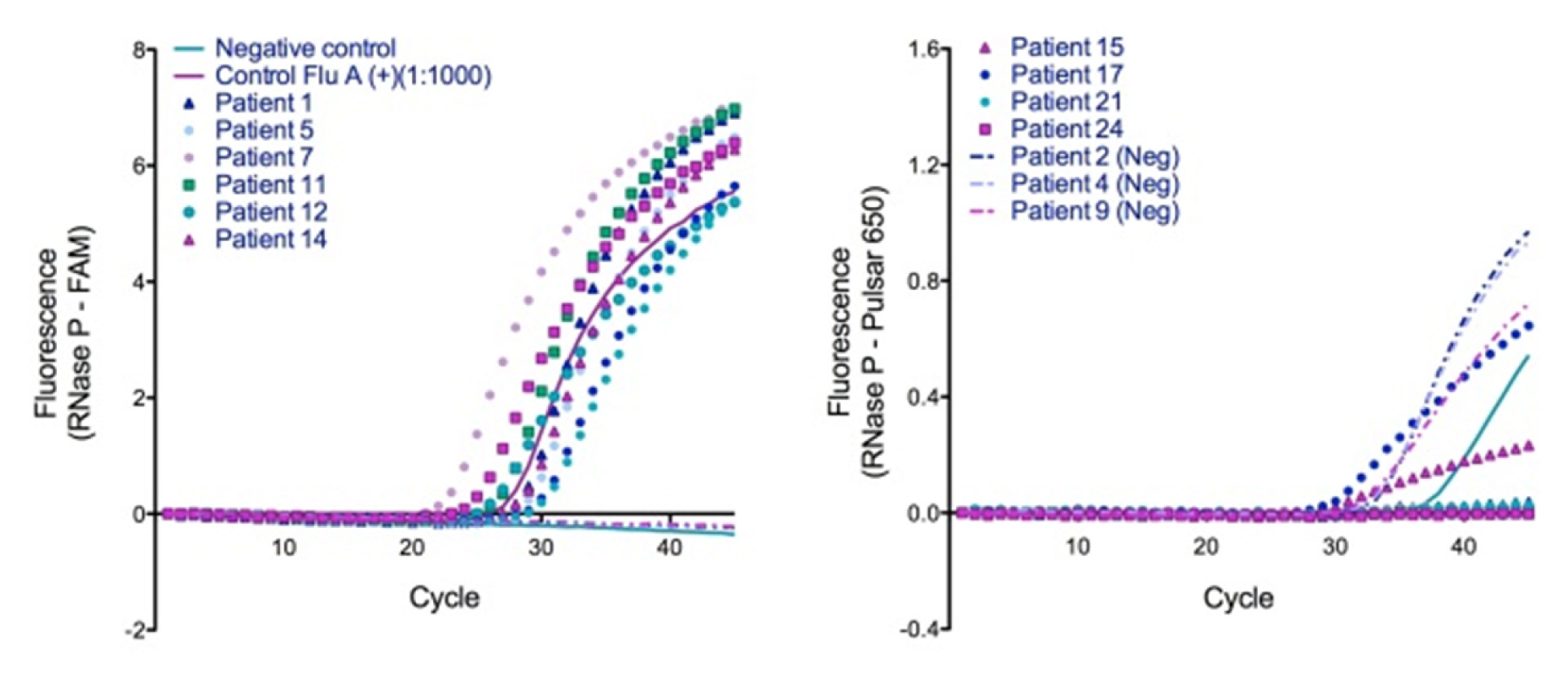
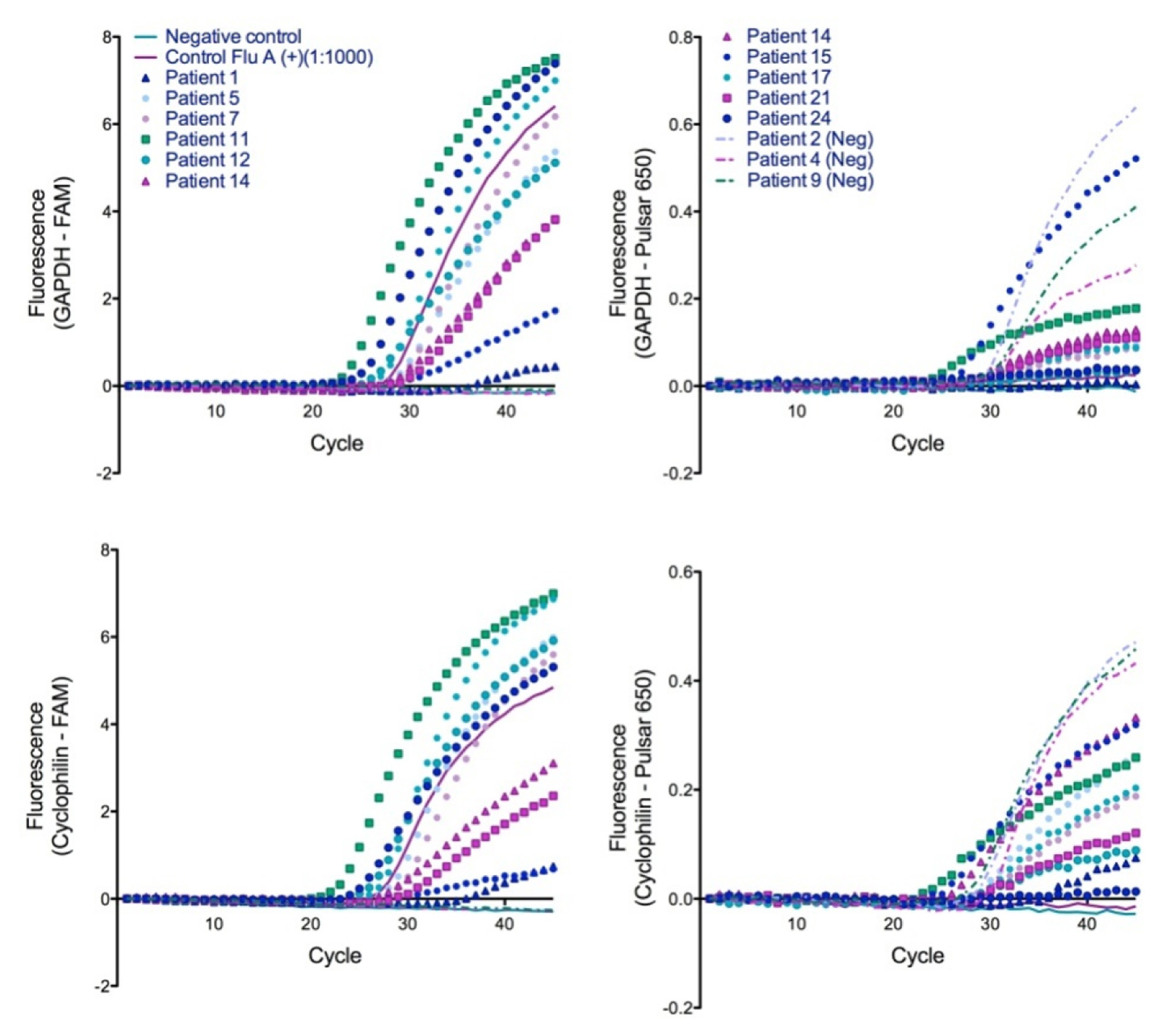
Consistent with the previously reported duplex assay using a single target, our dual-target duplex assays were successful in detecting infection in clinical samples. Using hRP, GAPDH, and cyclophilin as ICs, we were able to confirm the duplex assay using flu positive nasal wash samples [Table/Fig-2&3]. Both the ICs and viral target were RNA-based, requiring a reverse transcription (RT) step prior to PCR and it is unknown how this RT step would affecta DNA target assay. RNase inhibitors, which may have been needed for successful amplification of an RNA target from clinical samples [9], were not added. DNA target silencing in clinical matrices may be caused by PCR inhibitors present in the matrix [10].
Duplexed detection of a FAM-labeled influenza A probe (left) is possible with Pulsar®650 labeled IC targets based on 18S ribosomal RNA, GAPDH, RNase P, cyclophilin, gyrase and β-actin. Strong amplification of the IC probes (right) was observed for 18S RNA, GAPDH, and cyclophilin, whereas for little to no amplification was seen for the others
Demonstration of single-tube duplexed RT-PCR assay for clinical detection of influenza in nasal wash using RNase P as the internal control. Left, FAM labeled FluA probe; Right, Pulsar®650 labeled IC probe. Legend is shared between panels. (Neg) = clinically negative patient samples
Demonstration of single-tube duplexed RT-PCR assay for clinical detection of influenza in nasal wash using GAPDH (top) and Cyclophilin (bottom) as the internal control. Left, FAM labeledFluA probe; Right, Pulsar®-650 labeled IC probe. Legend is shared between panels. (Neg) = clinically negative patient samples
Conclusion
We have presented a novel method of performing duplex assays targeting influenza A with a human targeted IC on LC 1.5 technology. This duplex assay employed a probe utilizing the new Biosearch Technologies fluorophore Pulsar® 650, which is capable of fluorescence emission far from the excitation wavelength, resulting in spectra that can be read on channel 3 of the LC 1.5, or channel 6 of the LC 2.0. The potential of the duplexed assay to simultaneously detect DNA and RNA is exciting and could save sample, reagents, and time; and increase the overall throughput of the LightCycler for clinical detection.
[1]. Jothikumar P, Hill V, Narayanan J, Design of FRET-TaqMan Probes for Multiplex Real-Time PCR Using an Internal Positive ControlBioTechniques 2009 46:519-24. [Google Scholar]
[2]. Probert WS, Ely J, Shrader K, Atwell J, Nossoff A, Kwan S, Identification and evaluation of new target sequences for specific detection of Bordetella pertussis by real-time PCRJ. Clin. Microbiol 2008 46(10):3228-31. [Google Scholar]
[3]. World Health Organization. CDC Protocol of real-time RT-PCR for swine influenza (H1N1). Atlanta, GA: WHO Collaborating Centre for Influenza at CDC; 2009 [Google Scholar]
[4]. Niki T, Iba S, Tokunou M, Yamada T, Matsuno Y, Hirohashi S, Expression of vascular endothelial growth factors A, B, C, and D and their relationships to lymph node status in lung adenocarcinomaClin Cancer Res 2000 6:2431-39. [Google Scholar]
[5]. Taylor TB, Winn-Deen ES, Picozza E, Woudenberg TM, Albin M, Optimization of the performance of the polymerase chain reaction in silicon-based microstructuresNuc Acids Res 1997 25:3164-68. [Google Scholar]
[6]. Pasten C, Olave NC, Zhou L, Tabengwa EM, Wolkowicz PE, Grenett HE, Polyphenolsdownregulate PAI-1 gene expression in cultured human coronary artery endothelial cells: molecular contributor to cardiovascular protectionThrombosis Res 2007 121:59-65. [Google Scholar]
[7]. Zhang Z, Yamashita H, Toyama T, Omoto Y, Sugiura H, Hara Y, Quantitative determination, by real-time reverse transcription polymerase chain reaction, of aromatase mRNA in invasive ductal carcinoma of the breastBreast Cancer Res 2003 5:R250-56. [Google Scholar]
[8]. Goerke C, Bayer MG, Wolz C, Quantification of bacterial transcripts during infection using competitive reverse transcription-PCR (RT-PCR) and LightCycler RT-PCRClin. Diag. Lab Immunol 2001 8:279-82. [Google Scholar]
[9]. Fraga D, Meulia T, Fenster S, Real-time PCR. In: Gallagher SR, Wiley EA, editorsCurrent Protocols Essential Laboratory Techniques 2008 Hoboken, NJJohn Wiley & Sons:10.3.1-10.3.34. [Google Scholar]
[10]. Akane A, Matsubara K, Nakamura H, Takahashi S, Kimura K, Identification of the hemecompound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplificationJ Forensic Sci 1994 39:362-72. [Google Scholar]