Infantile Onset Alexander Disease with Normal Head Circumference: A Genetically Proven Case Report
Manisha Goyal1, Sumit Mehndiratta2, Mohammed Faruq3, Manish Kumar Dwivedi4, Seema Kapoor5
1 Senior Research Officer, Division of Genetics and Metabolism, Department of Pediatrics, Maulana Azad Medical College, New Delhi, India.
2 Junior Specialist, Department of Pediatrics, Maulana Azad Medical College, New Delhi, India.
3 Scientist, Genomics and Molecular Medicine, CSIR-Institute of Genomics and Integrative Biology, (IGIB-CSIR), Mall Road, Delhi, India.
4 Project Fellow, Genomics and Molecular Medicine, CSIR-Institute of Genomics and Integrative Biology, (IGIB-CSIR), Mall Road, Delhi, India.
5 Professor, Department of Pediatrics, Division of Genetics and Metabolism, Department Of Pediatrics, Maulana Azad Medical College, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Seema Kapoor, M-439, Ground Floor, Guruharkishan Nagar, Paschim Vihar, New Delhi-110063, India. Phone : 9968604313, E-mail : drseemakapoor@gmail.com
Alexander disease (AD) is an autosomal dominant leukodystrophy which predominantly affects infants and children. The infantile form comprises the most common form of AD. It presents before two years of age and characterized by macrocephaly, psychomotor regression, spasticity, pyramidal sign, ataxia and seizures. The diagnosis is based on magnetic resonance imaging (MRI) findings and confirmed by Glial fibrillary acidic protein (GFAP) gene molecular testing. We report an Indian case with normal head circumference.
Leukodystrophy, Macrocephaly, Psychomotor regression
Case Report
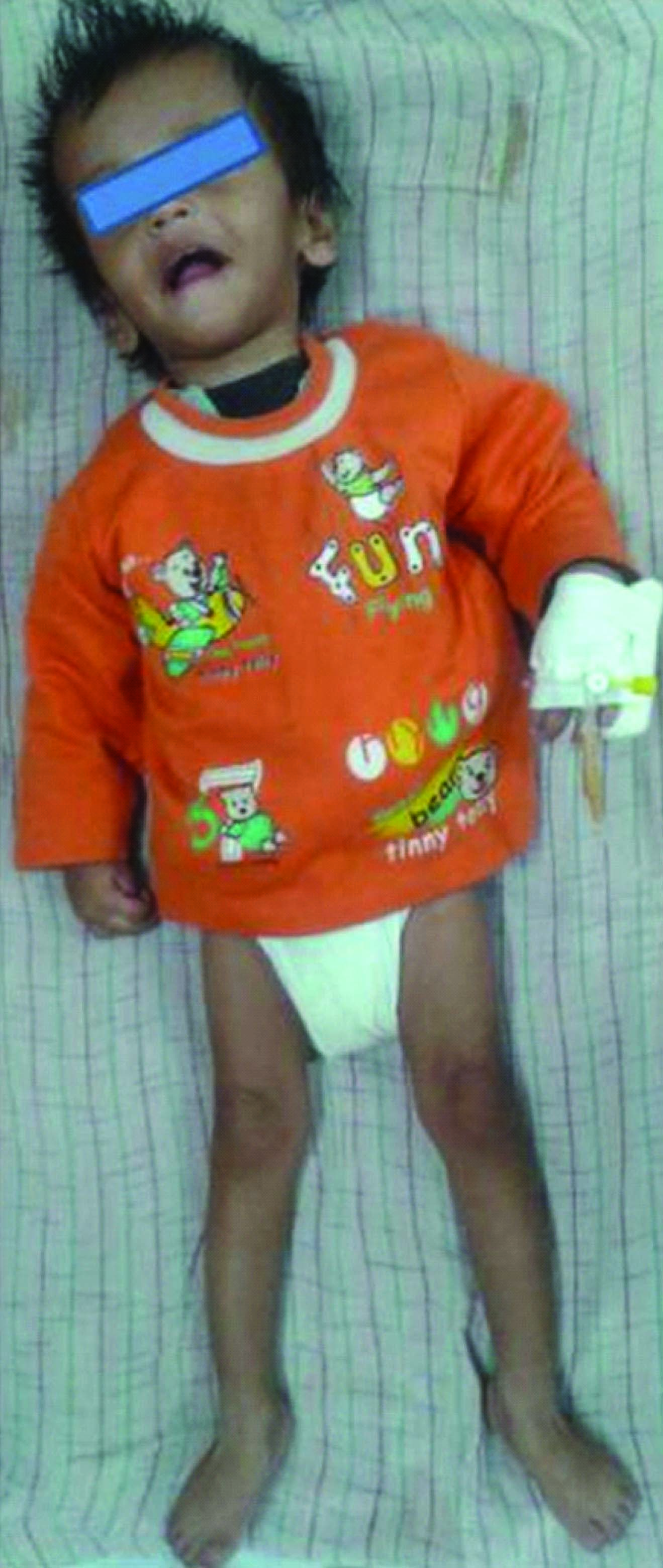
A 16-month-old boy presented to our genetic OPD for evaluation of developmental regression and seizures. He was second in birth order, born to non consanguineous couple at term after normal vaginal delivery with birth weight of 2.8 kg. Mother’s antenatal and perinatal history was uneventful. There was no history of any drug intake during pregnancy. He had attained social smile and neck holding at appropriate ages. He developed seizure at four months of age followed by significant regression in motor and cognitive skills whereby he lost neck holding and unable to recognise parents. Seizure was continuous and difficult to manage. Presently, at 16 monthss of age, he has partial neck holding, has no speech or interaction or recognition of parents. Parents gave a history of generalized spasticity of the body since six months of age. The patient has a six year old elder brother, intelligent, with normal development. There was no similar history in any family members from both sides of parents. On examination at 16 months of age, there were no dysmorphic features. Spasticity of upper and lower limbs with peripheral contractures at ankles was noted [Table/Fig-1]. His weight, length and head circumference were 7.5 kg (between -2 and -3 SD), 72 cm (between -2 to -3 SD) and 46 cm (between 0 to -1 SD), respectively. There was no hepato-splenomegaly or any neurocutaneous stigmata. His body tone was increased with intermittent tightening. Deep tendon reflexes were exaggerated in both upper and lower limbs with extensor planter at ankle joints.
16 months old boy with AD: note the spasticity and contracture at ankle joint,
Blood investigations including high performance liquid chromatography (HPLC) of serum amino acids, urine organic acid profile, serum ammonia and lactate level and thyroid function tests were within normal limit. Renal function tests, liver function test, hearing evaluation, ultrasonography abdomen were normal. Visual evoked potential (VEP) was subnormal with increased latencies. Magnetic resonance imaging (MRI) brain showed signal alteration of white matter, predominantely in the bilateral frontal regions also involving basal ganglia. There was hypointense line along the ventricular margin and areas of diffusion restriction in the bilateral lentiform nucleus and frontal white matter with cystic dilatation of cavum septum pellucidum [Table/Fig-2a&b]. Features were suggestive of Infantile form of Alexander disease (AD). Mutation analysis was advised. Consent was obtained from each of the participating members of the family in this study for genetic evaluation. A standard salting out protocol was followed for DNA isolation from the peripheral venous whole blood. The coding portion of the GFAP was amplified by polymerase chain reactions using nine set of primers. primer designing was done by Primer-3 software. The amplified products were subjected for Sanger method of sequencing using ABI-BigDye terminator sequencing chemistry and ABI3130xl capillary sequencer [1]. It revealed a de novo heterozygous mutation c.716G>A of p.Arg239His in the exon 4 of GFAPgene. Both the parents and elder brother were homozygous for wild type allele. The child was treated with anticonvulsant drugs.
Axial T1 and T2-weighted MRI images showing a periventricular rim (hyper intense in T1 and hypointense in T2 marked by single arrow) and signal alteration in periventricular white matter predominantly in frontal area (marked by double arrow)

He was admitted multiple times with uncontrolled seizures aggravated by fever associated with respiratory tract infection. During admission he was treated with antibiotics, antipyretic and intravenous anticonvulsant. Physiotherapy has been given.
Discussion
Alexander disease (AD): OMIM #203450 is an autosomal dominant leukodystrophy with frontal lobe preponderance that predominantly affects infants and children and usually results in death within ten years after onset. AD is characterized by progressive failure of central myelination and leukodystrophy on MRI and pathologically by the presence of Rosenthal fibers (eosinophilic cytoplasmic fusiform inclusions within astrocytic processes) adjacent to areas of demyelination. Rosenthal fibers are protein aggregates containing GFAP, ubiquitin, heat shock protein hsp-27, and B-crystallin [2]. Mutation in the GFAPgene encodes GFAP protein has been proven to cause AD [3,4]. Three forms are recognized according to the age of clinical presentations: infantile, juvenile, and adult. The infantile form comprises most common form (about 51% of reported cases) followed by juvenile (23%) and adult (24%) cases [5]. Singh et al., suggested neonatal form as subset of infants with neonatal onset (i.e., within 30 d of birth) [6]. Infantile AD presents in the first two years of life, typically with progressive psychomotor retardation with loss of developmental milestones, convulsions, macrocephaly, hyperreflexia and pyramidal signs, ataxia, hydrocephalus secondary to aqueductal stenosis, and predominant cerebral white matter abnormalities in the frontal lobe on brain MRI [7]. Juvenile patients have a slower clinical course (with bulbar signs, ataxia, and spasticity), and their intellectual abilities are usually preserved [5]. Adult patients have heterogeneous symptoms; some patients have relapsing- remitting neurological symptoms that mimic multiple sclerosis and are only diagnosed as AD during neuropathological examination. None of the patients with adult onset had macrocephaly, seizures, or cognitive defects [5].
Diagnosis in the present case was based on the presence of seizure, dystonic movements and MRI findings. The interesting feature in this case was the presence of normal head circumference despite of appearance of macrocephaly due to failure to thrive. He had poor weight gain because of poor feeding. Head circumference at birth and taken in between (9 months - 45.5 cm) were always normal. Although macrocephaly is reported as an essential feature of AD, we hereby report an infantile case of AD with normal head circumference. Gorospe et al., reported 12 genetically confirmed cases of AD including seven with infantile onset. All had megalencephaly at presentation [8]. Li et al., studied 26 infantile AD out of 44 patients with AD and found macrocephaly in 62 % cases. In their study seizures (92%) was the most common feature followed by cognitive defects (82%), bulbar signs (62%), ataxia (58%), and spasticity (52%) [4]. MRI findings of our case were compatible with AD. Van der Knaap et al., has been proposed specific MRI criteria for the diagnosis of AD including extensive, symmetric white matter abnormalities with frontal preponderance; periventricular signal changes; basal ganglia and thalamic signal changes; brainstem lesions; and contrast enhancement of multiple areas throughout the brain [9].
In our case mutation test revealed heterozygous mutation in exons 4 of GFAP gene carrying arginine mutations (p.Arg239His). It has been previously reported with AD patients and considered causative for AD due to its de novo appearance [1,3]. However, the patient’s parents and elder brother had a normal base sequence. Therefore, the R239L GFAP gene mutation in the present case is presumed to be a de novo mutation or gonadal mosaicism. GFAP mutations were detected in 93% of patients by amplification of exons 1, 4, and 8 in the study by Rodriguez et al., on the infantile type of AD [10]. Differential diagnosis of any infant with megalencephaly, developmental delay, spasticity, and seizures are organic acidurias, lysosomal storage disorders, peroxisomal biogenesis disorders, Zellweger syndrome spectrum. AD should be considered in the differential diagnosis of infantile leukoencephalopathy, even when no macrocephaly is present [11]. These conditions should be considered in the workup of AD. No specific therapy is currently available for AD. Management is supportive and includes nutritional care, antibiotic treatment for intercurrent infection, antiepileptic drugs for seizure control, physiotherapy and occupational therapy. Assessment is required for feeding/eating, digestive problems such as constipation and gastroesophageal reflux, growth measurements, psychological assessment for older patients, family and social structure to determine availability of adequate support system.
Conclusion
Analysis of the GFAP gene is a strong marker for infantile AD when diagnosed on the basis of clinical features and MRI findings even in the absence of macrocephaly.
[1]. Caroli F, Biancheri R, Seri M, Rossi A, Pessagno A, Bugiani M, GFAP mutations and polymorphisms in 13 unrelated Italian patients affected by Alexander diseaseClin Genet 2007 72:427-33. [Google Scholar]
[2]. Jacob J, Robertson NJ, Hilton DA, The clinicopathological spectrum of Rosenthal fibre encephalopathy and Alexander's disease: a case report and review of the literatureJ Neurol Neurosurg Psychiatry 2003 74:807-10. [Google Scholar]
[3]. Brenner M, Johnson AB, Boespflug-Tanguy O, Rodriguez D, Goldman JE, Messing A, Mutations in GFAP, encoding glial fibrillary acidic protein, are associated with Alexander diseaseNat Genet 2001 27:117-20. [Google Scholar]
[4]. Li R, Johnson AB, Salomons G, Goldman JE, Naidu S, Quinlan R, Glial fibrillary acidic protein mutations in infantile, juvenile, and adult forms of Alexander diseaseAnn Neurol 2005 57:310-26. [Google Scholar]
[5]. Gorospe JR, Alexander Disease. In: Pagon RA, Bird TD, Dolan CR, et al., editorsGeneReviews™ [Internet] 2002 Seattle (WA)University of Washington, SeattleNov 15, [Updated 2010 Apr 22]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1172 [Google Scholar]
[6]. Singh N, Bixby C, Etienne D, Tubbs RS, Loukas M, Alexander's disease: reassessment of a neonatal formChilds Nerv Syst 2012 28:2029-31. [Google Scholar]
[7]. Ashrafi MR, Tavasoli A, Aryani O, Alizadeh H, Houshmand M, Alexander Disease: Report of Two Unrelated Infantile Form Cases, Identified by GFAP Mutation Analysis andReview of Literature; The First Report from IranIran J Pediatr 2013 23:481-84. [Google Scholar]
[8]. Gorospe JR, Naidu S, Johnson AB, Puri V, Raymond GV, Jenkins SD, Molecular findings in symptomatic and pre- symptomatic Alexander disease patientsNeurology 2002 58:1494-500. [Google Scholar]
[9]. Van Der Knaap MS, Naidu S, Breiter SN, Blaser S, Stroink H, Springer S, Alexander disease: Diagnosis with MR imagingAJNR Am J Neuroradiol 2001 22:541-52. [Google Scholar]
[10]. Rodriguez D, Infantile Alexander Disease: Spectrum of GFAP Mutations and Genotype-phenotype correlationAm J. Hum Genet 2001 69:1134-40. [Google Scholar]
[11]. Nishri D, Edvardson S, Lev D, Leshinsky-Silver E, Ben-Sira L, Henneke M, Diagnosis by whole exome sequencing of atypical infantile onset Alexander disease masquerading as a mitochondrial disorderEur J Paediatr Neurol 2014 18:495-501. [Google Scholar]