Introduction
Cystic fibrosis (CF) is a life limiting multi systemic autosomal recessive genetic disorder whose spectrum varying from milder to severe phenotypes identified since the inclusion of this disease in new born screening programme of many countries. This disease occurs with frequency of 1 in 2500 newborns in Caucasians population [1]. Exact burden of disease is not known from India due to lack of facilities to diagnose this condition. First case of CF was reported early in 1968 [2]. Suggested incidence of CF in migrant Asians (Indians and Pakistanis) was deduced to be 1 in 10,800 to 12,000 [3,4]. Since then, there have been many publications citing diverse manifestations of disease from Indian subcontinent but prevalence based data is lacking. CF currently has no definitive cure. Mucoactive agents, bronchodialators and anti inflammatory medications are the mainstay of treatment but none of these alter the progression of the underlying disease. The life expectancy of patients with CF has risen steadily over the last 25y, from a median predicted age of survival of 25y in 1985 to 37y in 2011 [5]. Ivacaftor targets this disease at molecular level and is a treatise into the area of personalized medicine.
Understanding Pathophysiology
The CF gene on long(q) arm of chromosome 7 codes for a cAMP/PKA-dependent, ATP-requiring, membrane chloride ion channel, a member of the ATP binding cassete protein (ABC) family. This is of vital importance for the apical membranes of many secreting epithelia and known as CFTR (cystic fibrosis transmembrane conductance regulator) [6]. This gene encodes for a protein that helps transport water in and out of cell membrane throughout the body. The predominant role thereby is the production of thin and free flowing mucus which maintains an adequate airway surface liquid (ASL) and regulates epithelial sodium channels (ENaC). Other signals, such as those delivered by the vagal cholinergic input can produce some secretion but with a much increased viscosity.
In CF, defective protein leads to a reduction of CFTR-related chloride secretion which is associated with a secondary increase in epithelial sodium channel-mediated Na+ level at the expense of ASL caused by the absence of the inhibitory activity of the normal CFTR. The osmotic balance is altered such that fluid leaves the surface and moves into the submucosal space. Consequently, submucosal gland ducts are blocked by mucus, which also settles down on the cilia making them unable to function. Poor cilary function inturn leads to an increased propensity for bacterial infection. This is because ASL, not only contains ions but also glycoproteins, mucins, lactoferrin, defensins, lysozyme, IgA, antimicrobial surfactant proteins and secretory leukoprotease inhibitor, all contributing antimicrobial and immune defence mechanism.
There are currently over 1900 known mutations affecting CFTR [7]. CF mutations result mostly in impaired channel function, reduction in number or both at the cell surface. The most common mutation, ΔF508 (or Phe508del) deletes phenylalanine at amino acid position 508 and accounts for about 70% of CF alleles in Caucasian population; and approximately 90% of persons with CF carry at least one ΔF508 mutation [8]. This has been utilized to perform a second tier test in newborn screening for this disease. However, it is not predominant mutation in Asian CF patients. Studies from Indian CF patients found low frequency (19-31.1 %,) of del F 508 mutation [9,10]. Another mutation, G551D(or Gly551Asp or Celtic mutation), arises as a consequence of substitution of aspartate residue for a glycine residue at position 551 and accounts for 4 to 5% of alleles [8]. It abolishes ATP-dependent gating, resulting in an open probability that is approximately 100-fold lower than that of wild-type channels. The G551D mutation phenotype is associated with severe pancreatic insufficiency. The ΔF508 mutation causes CFTR to misfold, thereby disrupting its biosynthesis and delivery to the cell surface. This altered protein fails to be trafficked to the correct location in the cell and is generally destroyed by the proteasome. The small amount that does reach the correct location functions poorly. In contrast, G551D-CFTR channels reach the cell membrane but rarely open.
Six different classes of CTFR mutations: have been described [11]. Class I defects cause truncated protein translation, while class II defects result in misfolded CFTR. Both of these mutations result in impaired delivery of CFTR proteins to the cell surface. Class III and IV defects result in full-length CFTR that reach the cell surface but exhibit gating defects or reduced pore conductivity respectively. In addition, there are mutations that affect mRNA stability (Class 5) and CFTR stability (Class 6). Sometimes it is difficult to make a clear distinction between two or more of the classes of mutation, for example DF508 CFTR is poorly transported to the membrane and functions poorly compared to CFTR. Functional classification is the key to understanding the role of pharmacogenomics. [Table/Fig-1] depicts the classes of mutation based on their functional role along with the types of mutations.
Classes of mutation based on their functional role along with the types of mutations
| Class | Consequense of Mutation | Type of Mutation |
|---|
| I | No CFTR synthesis | Nonsense; Frameshift; Canonical splice |
| II | Failure of CFTR to traffic to the cell surface | Missense; Aminoacid deletion |
| III | Failure of CFTR to open at the cell surface | Missense; Aminoacid change |
| IV | Decrement in time, the CFTR channel is open at the cell surface | Missense; Aminoacid change |
| V | Decreased amount of CFTR at the cell surface | Splicing defect; Missense |
| VI | Unstable CFTR with decreased half-life at the cell surface | Missense; Aminoacid change |
Abbreviation: CFTR- CF transmembrane conductance regulator protein
Genotype- Phenotype Correlation: The CF phenotype is highly heterogeneous among individual patients, and even between siblings carrying identical CFTR mutations. Of the various clinical symptoms of CF, only pancreatic function has been shown to correlate well with CFTR genotype [12]. In particular, lung involvement in CF demonstrates poor genotype-phenotype correlation. Understandably, homozygous or compound heterozygous class I–III CFTR mutations are associated with an absence of or severely reduced production of functional CFTR, correlating well with a more severe phenotype. Class IV, V, and VI CFTR mutations are usually associated with some residual CFTR function and a more variable and sometimes milder disease
Approaches to Therapy: Since the discovery of genetic locus of this disease, scientists have tried various strategies to alter proteomics or modulate the mutation with a hope to offer a wider arena of pharmacotherapeutics. Ivacaftor is a role model in that direction. There are 3 types of pharmacological agents which can modulate the CTFR channels [13]. These include (i)Agents promoting trafficking of mutant CFTR to, and insertion into the membrane, and are called as correctors; ii) Agents which activate appropriately located mutant CFTR, thereby known as activators and iii) Agents that augment CTFR gating in response to CAMP signaling, known as potentiators. The term ‘opener’ was initially used instead of the term potentiators. However, potentiator is the more precise term since generally the channels need to be ‘primed’ before the potentiator can activate the channel. Indeed an opener per se may not be useful, leading to excessive activity. Ivacaftor is a prototype of the potentiator group.
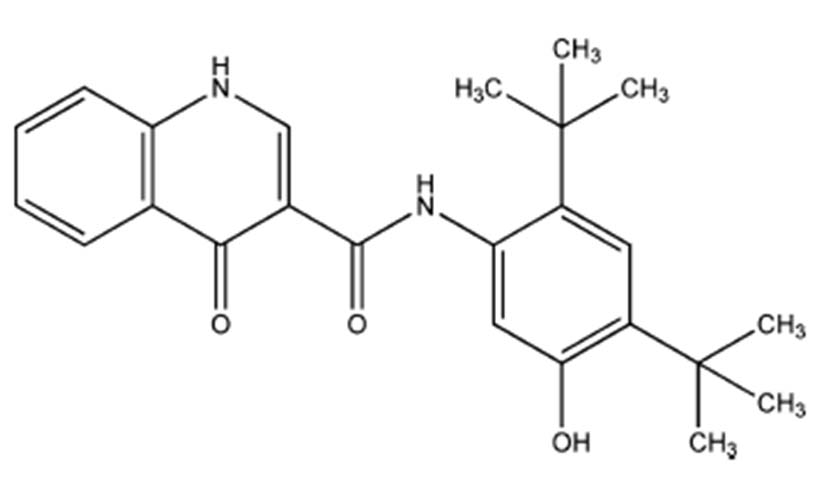
IVACAFTOR (VX-770) – Indications and Mechanism of Action : Ivacaftor (also known as Kalydeco or VX-770 prior to marketing for this indication) is chemically N-(2,4-Di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide [14] [Table/Fig-2].
Chemical structure of Ivacaftor
Mechanism of Action
Ivafactor is in principal increases CFTR channel opening. This property of opening the channels has been seen in both G551D mutation cells and delF508 mutation cells. Considering this utility dual therapy may tackle the disease present in the largest cohort of patients. Although, the exact mechanism by which ivacaftor opens the CFTR channel is not known, in vitro ivacaftor increases apical surface fluid and the movement of cilia. Further in vitro studies have elaborated on the mechanism further and have shown that ivacaftor opens the defective channel gate of mutant CFTR in a phosphorylation-dependent but ATP-independent manner [13].
Therapeutic Efficacy : Evidence from Previous Studies
The initial studies include a randomized, double-blind, placebo-controlled trial evaluating dose-ranging from 25–250 mg every 12h including 39 patients with CF [15]. On the basis of this multicentre phase II study, appropriate dosage of 150mg every 12h was selected for subsequent trials. Two subsequent trials involved two age groups, ages 6-11 y and those above 12y. The first one was a 48wk long randomised double blinded placebo controlled phase III trials called STRIVE (Age ≥ 12) and the second ENVISION for the same duration(Age 6-11 y).
Using 150mg twice daily regimen in addition to standard care improved lung function, pulmonary exacerbation index (STRIVE), body weight and health related quality of life. Beneficial effects were maintained upto 96 weeks post treatment in an ongoing open label extension (PERSIST) of the above programmes. Patients eligible for these trials had confirmed CF and the G551D mutation in at least one CFTR allele and were required to have an FEV1 40–90 % of predicted in STRIVE and 40–105 % of predicted in ENVISION [16,17]. The exclusion criteria of both studies included pulmonary exacerbation, acute respiratory infection, Mycobacterium abscessus, Burkholderia enocepacia or Burkholderia dolosa in sputum and changes in pulmonary disease therapy in the last 4wks. The primary endpoint of each trial was the absolute change from baseline in percent predicted FEV1 through week 24 of treatment.
Safety Profile: Four randomised control trials have been identified investigating the efficacy and safety of ivacaftor. The first study conducted by Accurso and colleagues evaluated the safety and adverse event profile of ivacaftor in patients age 18y or older with at least 1 G551D mutation. Doses in 25, 75, or 150 mg were studied for 14 d followed by a dose of 150 or 250 mg for 28 d [15]. It was concluded that ivacaftor was associated with very few severe side effects. Although efficacy was not a primary end point, an 8.7% (range, 2.3–31.3) change in forced expiratory volume in 1 sec (FEV1) from baseline was observed, and median sweat chloride decreased to 59.5 mmol/L. The dramatic shift occurred following the landmark global phase III study conducted by Ramsey and colleagues (STRIVE trial)[16] . They evaluated the efficacy and safety of ivacaftor 150mg twice daily for 48 wks in 161 patients age 12y or older with at least 1 G551D mutation. At 24wks, ivacaftor resulted in a 10.6% improvement in predicted FEV1 from baseline versus placebo. A 55% reduction in the risk of pulmonary exacerbations was observed. Mean values for sweat chloride at 24wks were 47.8 mmol/L in the ivacaftor group versus 100.0 mmol/L in the placebo group. Regarding the CF Questionnaire in evaluation of quality of life-Revised respiratory domain scores increased by 5.9 points versus 2.7 points in the placebo group by week 48 signifying improved quality of life. Patients in the ivacaftor group also gained 3.1 kg, which was also statistically significant when compared to placebo. In regards to safety, elevated hepatic enzyme levels led to discontinuation of ivacaftor in 1 subject with no effects in the other subjects.
Davies et al., (ENVISION trial) studied safety and efficacy of ivacaftor in children 6 to 11 y of age by a 48-wk study including 52 patients and resulted in a 12.5% improvement in FEV1 and 2.8kg weight gain compared to placebo [17]. Ivacaftor thus proved to be the first agent to show a reduction in the sweat chloride level to values below the diagnostic threshold for CF (60 mmol/L). Interestingly the decreases in sweat chloride did not correlate with improvement in FEV1 in those treated with ivacaftor. Ivacaftor also showed a better efficacy in young patients with l a mild form of the disease. Such findings suggest that treatment at an early stage may block or substantially slow down the progression of the disease.
Flume and colleagues evaluated the safety of ivacaftor 150mg twice daily for 96wks in patients age 12y or older with homozygous delta F508 CFTR mutations. Similar adverse events were reported between ivacaftor and placebo but efficacy in patients with this mutation was not demonstrable [18]. Understandably in patients without a gating defect its efficacy is doubtful.
Pharmacokinetics and Pharmacodynamics: Ivacaftor has good bioavailability orally and its absorption is enhanced with fat containing meals (20g of fat) which increases the bio availability of drug to 2 to 4 fold [19]. Peak plasma level is reached in 4h time and its half life is 12h. Majority of the drug is bound with albumin and alpha1 – acid glycoprotein and its elimination occurs via feces. It is metabolized by CYP3A to pharmacologically active M1 and inactive M6 metabolites.
The rate of ivacaftor absorption in children is similar to that in adults, although the predicted total body clearance of the drug in children is lower (e.g. 10 L/h in a 20kg child vs. 18.9 L/h in a 70kg adult) according to a population pharmacokinetic analysis, which may explain why exposure to ivacaftor was higher in children than in adults in phase II and III studies.
Elimination via enterohepatic circulation necessitates dosage adjustment according to severity of hepatic impairment. In patients with severe and moderate hepatic dysfunction (Child- Pugh class C), dose should be reduced to 150mg daily or less frequently respectively. Dose should be reduced to 150mg daily with moderate hepatic dysfunction (Child- Pugh class B). No dosage modification is required in patients with renal impairment except for severe (creatinine clearance of less than 30 ml/min) and end stage renal disease.
Dosing: Patients with an unknown genotype should have the presence of the G551D mutation confirmed before being treated with ivacaftor. The approved dose is 150mg twice daily for adults and children alike [19]. One should exclude any CYP3A4 modifying medications or foods prior to starting ivacaftor.
Adverse Effects: Its safety profile is good and adherence/compliance to the drug is high at 90%, which compares favourably with adherence rates reported for some other CF therapies, such as inhaled antibiotics. The adverse effects that occur more frequently in the ivacaftor group are headache, upper respiratory tract infections , nasal congestion , rash and dizziness. None of these are serious to lead to discontinuation. Patients with CF often have moderate elevations in levels of AST or ALT and transaminase elevations have, overall, occurred with similar incidence with ivacaftor as with placebo in clinical studies, they have been documented more frequently with ivacaftor group. Thus, it is recommended that AST and ALT or hepatic function should be assessed before starting treatment with ivacaftor, every three months during the initial year of therapy and then once every year thereafter. If transaminase levels increase during treatment, patients should be monitored closely until the abnormalities resolve.However, although safety data are available for 60 weeks of treatment in the trials, the long-term safety profile of ivacaftor over a lifetime or its safety in young children is unknown. Most recently, an animal study showed that juvenile rats treated with ivacaftor from postnatal days 7–35 at dose levels of 10 mg/kg/day and higher (approximately one tenth the maximum recommended human dose) developed cataracts [20].
Drug Interactions: Ivacaftor is a weak inhibitor of CYP3A, P glycoprotein, CYP2C8, and CYP2C9. Co-administration of Ivafactor with strong CYP3A inducers such as rifampin, Phenobarbital, phenytoin, carbamazepine is not recommended [19]. Co-adminstration of Ivacaftor with strong CYP3A inhibitors requires the dose of ivacaftor to be reduced to 150mg orally twice a week. Whereas, it is reduced to 150mg orally daily, when it administered with moderate CYP3A inhibitors [19]. Thus co- administration with CYP3A inducers needs careful adjustment of drug dosages depending upon degree of inhibition. Foods containing CYP3A inhibitors such as grapefruit should be avoided as they may increase blood levels of ivacaftor. CYP2C9 may be inhibited by ivacaftor, necessitating monitoring of the international normalized ratio if the drug is coadministered with the CYP2C9 substrate warfarin.
Risk Stratification During Pregnancy and Lactation: In animal studies no evidence of tumorigenicity was observed. Impaired fertility was found in male and female rats at doses of 200 mg/kg/day. These impairments are attributed to severe toxicity as none were observed at doses less than or equal to 100 mg/kg/day. Ivacaftor belongs to pregnancy category B and may be excreted in breast milk. Currently there is no data available on human breast milk excretion and caution should be exercised if administered to nursing women [21].
Pharmaco-Economic Considerations: The cost of ivacaftor (150mg twice daily for one year) is 294,000 $, which may be prohibitive for patients, but it is not totally out of line with prices for other drugs for rare diseases [22]. Approval for use: It has been approved by both US and UK FDA for use in patients with CF carrying G551D mutation.
Alterntive Approaches And Potential Add On`s: This communiqué would be incomplete if other mutation modulating drugs are not mentioned. The first drug deserving mention is Ataluren (a CTFR suppressor) for patients with class I nonsense mutation and is under phase I trials. This drug works in part by binding the ribosome to allow the protein synthesis machinery to read through the premature stop signals in the RNA code. It is also suggested to be useful for Duchene muscular dystrophy [23].
The second group of drugs are the activators of alternative chloride channels – For this action it is desirable that the channels should be located apically so that treatment by aerosol is possible and it is essential that the signal transduction pathways are independent of cAMP metabolism. It was established that both purine (ATP) and pyrimidine (UTP) triphosphate nucleotides met the second criterion and were able to activate P2Y2 receptors located on the apical membranes of airway epithelial cells. When applied to either normal or CF airway epithelia, these agents increase chloride secretion via increases in ionized calcium that secondarily activate calcium-activated chloride channels (CaCC) present in the apical membrane. UTP analogues were chosen for development as metabolism of ATP by ectonucleotidases give rise to ADP, AMP and adenosine, all of which can cause bronchoconstriction in some circumstances. A further bonus of developing P2Y2 agonists for potential clinical use was the realization that they also inhibit electrogenic sodium absorption in both wild-type and DF508 CFTR expressing human bronchial epithelia (e.g., Denufusol tetrasodium (INS37217) or lancovutide). The nonapeptide antibiotic duramycin is supposed to cause calcium release from intracellular stores and stimulates calcium influx (Cloutier et al.,) thus activating CaCC [24].
The chloride channel type 2 (ClC-2) is widely distributed in epithelia and can contribute to native chloride secretion. Lubiprostone, a putative ClC-2 channel opener, apparently causes non-PKA dependent chloride secretion [25]. Another set of drugs are those reducing ENaC activity-The highly selective ENaC blocking drug amiloride was used in a clinical trial 25 y ago in an attempt to prevent volume depletion of the ASL in CF patients but no therapeutic improvement was established. The reason appeared to be the low potency and the rapid clearance of amiloride from the lung. The benzyl derivative of amiloride was shown to be an order of magnitude more effective than the parent compound and had a longer duration of effect [26].
Airway Rehydrators - Hypertonic saline and mannitol - There have been a number of clinical trials where improvements in lung function in CF patients have been recorded, for example following inhalation of 4 mL of 7% saline, twice daily. In an attempt to produce a greater osmotic gradient that dissipates more slowly attention turned to mannitol. Inhalation of dry mannitol powder (420mg), twice daily for 2 wks improved lung function in CF patients and was well tolerated [27].
Dornase alfa- Vibrating sinonasal inhalation of dornase alfa reduces rhinosinusitis symptoms in CF patients [28].
Inhaled Tobramycin - Aminoglycoside antibiotics, such as G-418, tobramycin and gentamicin, allow translational read through allowing it to proceed to the end of the transcript and correction of nasal PDs with gentamicin has been shown in CF patients with stop mutations [29].
Gene Therapy - No clinical benefit has thus far been recorded. Access of the transfecting agents to the surface epithelial cells is hampered by the overlying mucus while transfection of the submucosal glands poses special problems. Humans carrying only one CF gene do not have symptoms of CF, and this together with other evidence that indicates as little as 10% normal CFTR may be sufficient, makes DF508 a major target for drug therapy, especially since it would benefit a majority of patients. It has been known for many years that mammalian cells expressing DF508 CFTR revert to wild type when the temperature is lowered [30]. Obviously a chemical chaperone that could achieve the same result would be of particular interest for patients with a DF508 mutation.
What Next?
There occurs many questions to be unanswered; is it safe in infants and children? Is it prudent to give it to those newborns that are identified through newborn screening programme of various countries? Can we prevent involvement of lung and liver in G551D-CFTR mutation carrying patients by offering this magic bullet [31]. These are questions which can be answered by only further ongoing research in this field
First, we cannot quantitatively assay CFTR function in vivo. The nasal potential difference and sweat chloride concentration provide little quantitative information about CFTR chloride permeability and involve other complex transport processes. Moreover, their relationship to CFTR activity is nonlinear. The relationship between CFTR function and disease could differ in the lung, pancreas, and Liver thus further limiting our ability in assessing CTFR function in vivo [32].
At this time, ivacaftor has only been studied as adjunct therapy to standard of care. Whether use of ivacaftor could enable some standard care medications to be discontinued remains to be determined.
Conclusion
There is a flurry of activity in clinical trials with therapies aimed at proximal events in CF pathophysiology. It is yet to be seen whether these newer medicines will prove of some sustained benefit to our patients and reduce their sufferings.
The introduction of this new drug has opened the window of hope for G551D mutation CF patients. It is seemingly well tolerated and offers simplicity in dosing. In addition to gating mutations, the efficacy and safety of ivacaftor has also been evaluated in patients homozygous for the class II mutation F508del, the most common mutation. Definitely, the introduction of this drug has ushered in the era of personalized pharmacotherapeutics.
Until some additional questions above are answered, the CF community should proceed with a combination of hope and caution.
Abbreviation: CFTR- CF transmembrane conductance regulator protein
[1]. Ratjen F, Doring G, CFLancet 2003 36:681-89. [Google Scholar]
[2]. Bhakoo ON, Kumar R, Walia BN, Mucoviscidosis of the lung. Report of a caseIndianJournal of Pediatrics 1968 35:183-85. [Google Scholar]
[3]. Goodchild MC, Insley J, Rushton DI, Gaze H, CF in 3 Pakistani childrenArchives of Disease in Childhood 1974 49:739-41. [Google Scholar]
[4]. Powers CA, Potter EM, Wessel HU, Lloyd-Still JD, CF in Asian IndiansArchives of Pediatrics & Adolescent Medicine 1996 150(5):554-55. [Google Scholar]
[5]. Bethesda M. Patient Registry. Annual Data Report. Available from: http://www.cff.org/LivingWithCF/QualityImprovement/PatientRegistryReport/: CF Foundation; 2011[Accessed 5 feb 2013] [Google Scholar]
[6]. Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Identification of the CF gene: cloning and characterization of complementary DNAScience 1989 245:1066-73. [Google Scholar]
[7]. CF Mutation Database. CFTR1, 2013. Available from: http://www.genet.sickkids.on.ca/cftr/StatisticsPage.html. 2013[Accessed 5 feb 2013] [Google Scholar]
[8]. Trust CFRR. Annual Data Report 2012 . Available from: http://www.cftrust.org.uk/aboutcf/publications/cfregistryreports/UK_CF_Registry_-_Annual_Data_Report_2012.pdf. [Accessed Feb 2014] [Google Scholar]
[9]. Sachdeva K, Saxena R, Puri R, Bijarnia S, Kohli S, Verma IC, Mutation analysis of the CFTR gene in 225 children: identification of five novel severe and seven reported severe mutationsGenetic Testing and Molecular Biomarkers 2012 16:798-801. [Google Scholar]
[10]. Shastri SS, Kabra M, Kabra SK, Pandey RM, Menon PS, Characterisation of mutations and genotype-phenotype correlation in CF: experience from IndiaJournal of CF : official journal of the European CF Society 2008 7:110-15. [Google Scholar]
[11]. Welsh MJ, Smith AE, Molecular mechanisms of CFTR chloride channel dysfunction in CFCell 1993 73(7):1251-54. [Google Scholar]
[12]. Davis PB, Schluchter MD, Konstan MW, Relation of sweat chloride concentration to severity of lung disease in CFPediatric Pulmonology 2004 38:204-09. [Google Scholar]
[13]. Derichs N, Targeting a genetic defect: CF transmembrane conductance regulator modulators in CFEur Respir Rev 2013 22:58-65. [Google Scholar]
[14]. Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770Proc Natl Acad Sci U S A 2009 106:18825-30. [Google Scholar]
[15]. Accurso FJ, Rowe SM, Clancy JP, Boyle MP, Dunitz JM, Durie PR, Effect of VX-770 in persons with CF and the G551D-CFTR mutationThe New England Journal of Medicine 2010 363:1991-2003. [Google Scholar]
[16]. Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Drevinek P, A CFTR potentiator in patients with CF and the G551D mutationThe New England Journal of Medicine 2011 365:1663-72. [Google Scholar]
[17]. Davies JC, Wainwright CE, Canny GJ, Chilvers MA, Howenstine MS, Munck A, Efficacy and safety of ivacaftor in patients aged 6 to 11 years with CF with a G551D mutationAmerican journal of Respiratory and Critical Care Medicine 2013 187:1219-25. [Google Scholar]
[18]. Flume PA, Liou TG, Borowitz DS, Li H, Yen K, Ordonez CL, Ivacaftor in subjects with CF who are homozygous for the F508del-CFTR mutationChest 2012 142:718-24. [Google Scholar]
[19]. Robertson SM, Luo X, Dubey N, Li C, Chavan AB, Gilmartin GS, Clinical drug-drug interaction assessment of ivacaftor as a potential inhibitor of cytochrome P450 and P-glycoproteinJ Clin Pharmacol 2014 Aug 7 doi: 10.1002/jcph.377. [Epub ahead of print] [Google Scholar]
[20]. Administration USFDA. Notification to the CF Community on Kalydeco (ivacaftor). Available from: http://www.fda.gov/Drugs/DrugSafety. [Accessed 5 September, 2013] [Google Scholar]
[21]. Condren ME, Bradshaw MD, Ivacaftor: a novel gene-based therapeutic approach for CFJ Pediatr Pharmacol Ther 2013 18:8-13. [Google Scholar]
[22]. Kaiser J, Personalized medicine. New CF drug offers hope, at a priceScience 2012 335:645 [Google Scholar]
[23]. Loudon JA, Ataluren: a ‘no-nonsense’ approach for pulmonary diseasesPulmonary pharmacology & therapeutics 2013 26:398-99. [Google Scholar]
[24]. Cloutier MM, Guernsey L, Sha’afi RI, Duramycin increases intracellular calcium in airway epitheliumMembr Biochem 1993 10:107-18. [Google Scholar]
[25]. Mohammad-Panah R, Ackerley C, Rommens J, Choudhury M, Wang Y, Bear CE, The chloride channel ClC-4 co-localizes with CF transmembrane conductance regulator and may mediate chloride flux across the apical membrane of intestinal epitheliaThe Journal of Biological Chemistry 2002 277:566-74. [Google Scholar]
[26]. Cuthbert AW, Shum WK, Characteristics of the entry process for sodium in transporting epithelia as revealed with amilorideThe Journal of physiology 1976 255:587-604. [Google Scholar]
[27]. Daviskas E, Anderson SD, Jaques A, Charlton B, Inhaled mannitol improves the hydration and surface properties of sputum in patients with CFChest 2010 137:861-68. [Google Scholar]
[28]. Mainz JG, Schien C, Schiller I, Schädlich K, Koitschev A, Koitschev C, Sinonasal inhalation of dornase alfa administered by vibrating aerosol to CF patients: A double-blind placebo-controlled cross-over trialJ Cyst Fibros 2014 Mar 1 pii: S1569-1993(14):00045-9.doi: 10.1016/j.jcf.2014.02.005. [Epub ahead of print] [Google Scholar]
[29]. Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Gentamicin-induced correction of CFTR function in patients with CF and CFTR stop mutationsThe New England Journal of Medicine 2003 349:1433-41. [Google Scholar]
[30]. Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ, Processing of mutant CF transmembrane conductance regulator is temperature-sensitiveNature 1992 358:761-64. [Google Scholar]
[31]. Davis PB, Therapy for CF—the end of the beginning?N Engl J Med 2011 365:1734-35. [Google Scholar]
[32]. Welsh MJ, Targeting the basic defect in CFN Engl J Med 2010 363:2056-57. [Google Scholar]