Molecular Docking Study for Inhibitors of Aggregatibacter actinomycetamcomitans Toxins in Treatment of Aggressive Perioodontitis
Makesh Raj L.S.1, Jude J.2, Kannan I.3, Sai Krishna P.4, Shankar K.A.5
1 Reader, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Tagore Medical College and Hospital, Rathinamangalam, Chennai, Tamil Nadu, India.
2 Senior Lecturer, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Tagore Medical College and Hospital, Rathinamangalam, Chennai, Tamil Nadu, India.
3 Assistant Professor, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Tagore Medical College and Hospital, Rathinamangalam, Chennai, Tamil Nadu, India.
4 Professor, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Tagore Medical College and Hospital, Rathinamangalam, Chennai, Tamil Nadu, India.
5 Reader, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Tagore Medical College and Hospital, Rathinamangalam, Chennai, Tamil Nadu, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Makesh Raj, Reader, Department of Oral Maxillofacial and Pathology, Tagore Dental College and Hospital, Rathinamangalam, Chennai, Tamil Nadu - 600 127, India. Phone : 91 9791071687, E-mail : makeshraj1981@gmail.com
Introduction: Periodontitis is a chronic inflammatory disease of the periodontal tissues causing periodontal attachment loss and destruction of the alveolar bone which leads to mobility and loss of teeth. Aggregatibacter actinomycetemcomitans (Aa) is a gram negative, capnophilic, coccobacillus that plays an important role in aggressive Periodontitis. Aa produces a variety of virulence factors that facilitate the colonization, invasion and destruction of the periodontal tissues. Leukotoxin and cytolethal distending toxin (Cdt) are most important virulence factors of Aa.
Materials and Methods: The three dimensional structure of leukotoxin was derived by Easy modeller software and Cdt was retrieved from RCSB database. The possible binding sites of toxins were searched using binding site prediction tool Q site finder. A total of 1000 ligands of flavanol derivatives were generated with the help of software ACD chemsketch. Rapid virtual screenings of these compounds were performed in the docking tool iGEMDOCK v2.0. Based on the binding energy, six ligands were selected for the further study. The selected six ligands were then analysed for drug relevant properties based on “Lipinski’s rule of five” and other drug like properties. The accurate docking of six ligands was performed using docking tool iGEMDOCK v2.0.
Results: From the present study, it has been found that carboxyl {(2R,3R)-3,7 dihydroxy 4-oxo-2(3,4,5-trihydroxyphenyl)-3,4-dihydro2H-chromen-5-yl} oxonium, which is a novel compound can effectively act as an inhibitor for both the toxins.
Conclusion: The leucotoxin and cytolethal distending toxin of Aa is found to be the major virulence factors involved in the causation of aggressive periodontitis. Hence the inhibitors of these toxins can be an effective drug in treatment of aggressive periodontitis.
Aggressive periodontitis, Aggregatibacter actinomycetamcomitans, Cytolethal distending toxin, Molecular docking, Leucotoxin
Introduction
Periodontal disease is probably the most common chronic infectious disease of humans and important global health issue. Most common type is chronic Periodontitis which progresses slowly, while the aggressive form shows rapid tissue loss and often occurs in younger patients. Aggressive Perioodontitis (Ap) is defined by the International Workshop for Classification of Periodontal Diseases and Conditions in 1999 as “multifactorial, severe and rapidly progressive form of Periodontitis, which primarily but not exclusively affects younger patients”.
The characteristic features of the Ap are, amounts of microbial deposits are inconsistent with the severity of periodontal tissue destruction. Elevated proportion of Actinobacillus actinomycetemcomitans (Aa) and in some populations Porphyromonas gingivalis. Phagocytic abnormalities seen in the patients affected by aggressive Periodontitis. Hyper-responsive macrophage phenotype including elevated levels of PGE2 and IL-1β are also evident [1,2].
Aa plays a major role in pathogenesis of aggressive peridontitis. Aa produces a variety of virulence factors (toxins) that facilitate the colonization, invasion of micro organisms and destruction of the periodontal tissues. The various virulence factors of Aa are leucotoxin, cytolethal distending toxin, lipopolysaccharides, Surface-associated material, chemotactic inhibition factors, proteases, collagenases etc [3].
Of these, leucotoxin and cytolethal distending toxin (Cdt) are the most important virulence factors expressed by Aa to cause aggressive Periodontitis. Leucotoxin is a family of pore forming toxins that destroys leukocytes by two mechanisms namely membranolytic activity producing pores in the target cell and rapid influx of Ca2+ into the cell leading to necrosis and apoptosis. Once the leukocytes are destroyed it is easy for the micro organism to invade the host system.
Cytolethal distending toxin (Cdt) induces apoptosis of lymphocytes by DNAse activity through caspase and also inhibits proliferation of fibroblasts and periodontal ligament cells. There by reducing the defense mechanism and regenerative capacity of the host [4–7]. Thus, production of effective inhibitors of leucotoxin and Cdt can be an effective drug in the prevention and treatment of aggressive peridontitis caused by Aa.
In-silico methods is a computer based method, which was developed and widely applied in the field of pharmacology to discover and optimization of novel molecules with affinity to a target, the clarification of absorption, distribution, metabolism, excretion and toxicity properties as well as physicochemical characterization. In silico method includes database searching, quantitative structure-activity relationships, similarity searching, pharmacophore identification, computational modeling and docking.
In the present study, an attempt has been made to design an effective inhibitor (a novel drug) againts leucotoxin and Cdt using insilico docking method which can be used to prevent aggressive peridontitis.
Materials and Methods
Protein preparation
Obtaining three dimensional structure of the toxins is the first step in the process of insilico docking. We attempted to obtain the three dimensional structure of the Cdt and leukotoxin.
The three dimensional structure of Cdt was retrieved from RCSB-PDB data base (PDB code 2F2F) [Table/Fig-1] But three dimensional structure of leucotoxin was not available in database. So it was necessary for us to derive the three dimensional structure of leukotoxin. Its structure was derived by following method:

Protein sequence of leucotoxin was obtained as FASTA sequence from UNIPROT database. (In bioinformatics, FASTA format is a text-based format for representing either nucleotide sequences or peptide sequences, in which nucleotides or amino acids are represented using single-letter codes)
Protein sequence similar to leucotoxin was obtained by BLASTP in UNIPROTKB. In the present case hemolysin of E. coli has the FASTA sequence similar to that of leukotoxin.
3D structure of hemolysin is retrieved from RCSB database.
Finally, 3D structure of leucotoxin is obtained with help of 3D structure of hemolysin using EASYMODELLER (HOMOLOGY MODELLING) [Table/Fig-2,3] (MODELLER is a computer program that models three-dimensional structures of proteins and their assemblies by satisfaction of spatial restraints)
Derived structure of Leukotoxin

FASTA sequence for leukotoxin

Active site prediction
Identifying the location of ligand binding sites on a protein is of fundamental important in molecular docking. Q-Site Finder is a new energy-based method for predicting protein-ligand binding sites. The method analyses interaction energies of a methyl probe with a protein using software developed by Jackson. The possible active binding sites of toxins were obtained using online binding site prediction tool Q site finder [8]. The binding sites which are more flexible were selected for this study.
Generation and optimization of Ligand
A total of 500 ligands of flavanol derivatives in 2D format were generated with software ACD chemsketch [9]. The ligands were first saved in mol format, and then converted to pdb format with help of OPEN BABEL software (www.vcclab.org/lab/babel/start.html). Rapid virtual screening of these compounds was performed in the docking tool iGEMDOCK v2.0[10]. A population size of 150 is set with 70 generation and one solution for quick docking. Based on the binding energy, a total of six ligands were selected for the further study. The selected six ligands were then analysed for drug- relevant properties based on “Lipinski’s rule of five”. Other drug like properties was analysed using OSIRIS Property Explorer (http://www.organicchemistry.org/prog/peo/) and molecular property explorer (http://www.molsoft.com/mprop/). On the basis of binding affinity and drug like properties, all these six ligands were taken for further molecular docking study.
Protein-ligand docking
iGEMDOCK is an integrated tool that creates virtual screening environment from preparations through post-screening analysis with pharmacological interactions. First, iGEMDOCK provides interactive interfaces to prepare both the binding site of the target protein and the screening compound library. Then, each compound in the library is docked into the bindingsite by using the docking tool iGEMDOCK [Table/Fig-4,5] Subsequently, iGEMDOCK generates protein-compound interaction profiles of electrostatic, hydrogen-bonding, and van der Waals interactions [Table/Fig-6] Based on these profiles and compound structures, iGEMDOCK infers the pharmacological interactions and clusters the screening compounds for the post-screening analysis. Finally, iGEMDOCK ranks and visualizes the screening compounds by combining the pharmacological interactions and energy-based scoring function of iGEMDOCK. The selected six ligands were subjected to accurate docking (very slow docking) by setting population size of 800 is set with 80 generation and 10 solutions. After the completion of the docking, the post docking analysis was performed to find the docking pose and its energy values.

Docking site for leukotoxin
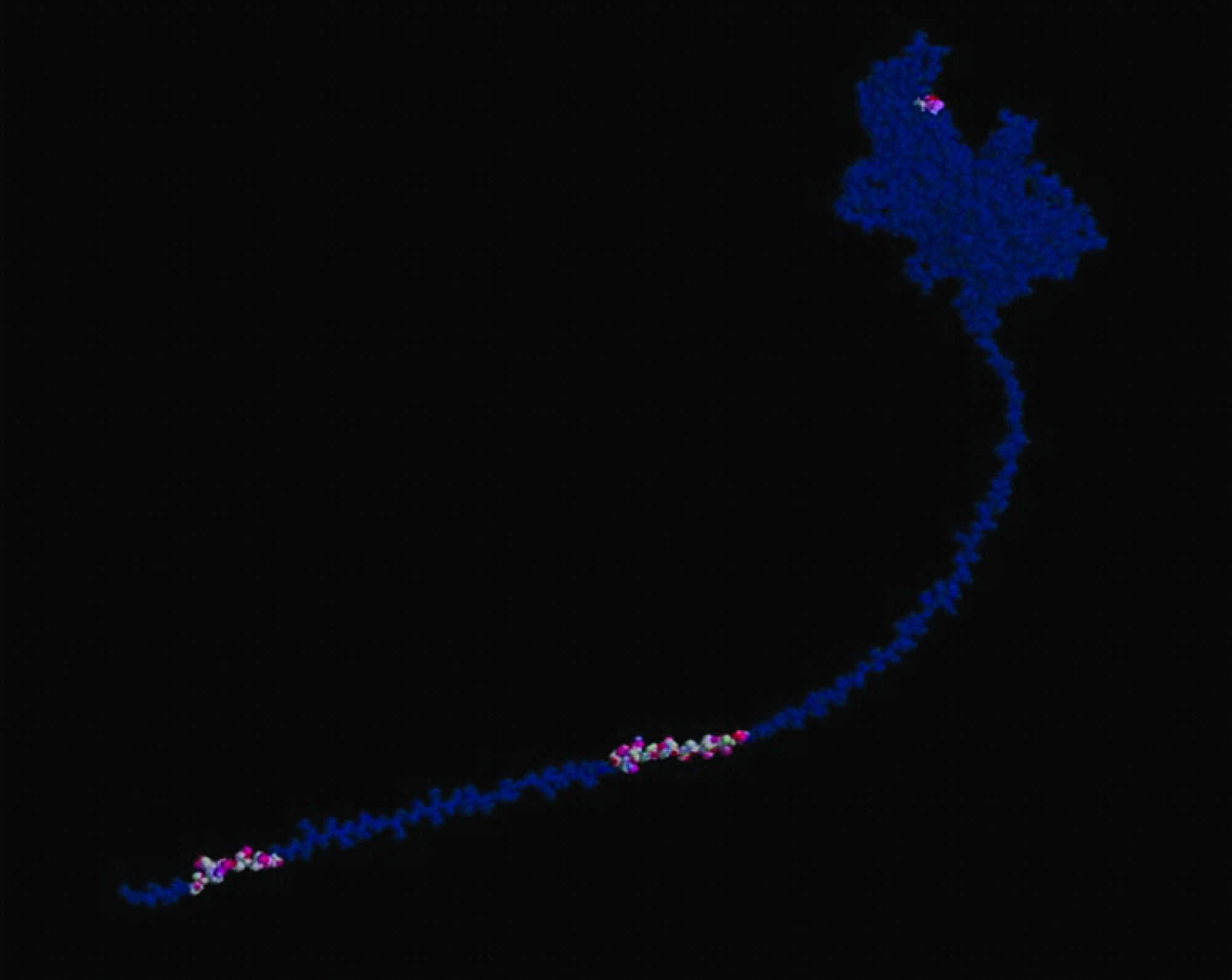
Protein-compound interaction profiles of ligands
| s.no | Ligand | Total binding energy | Vanderwaals force | Hbond | Electrostatic bond |
|---|
| 1 | (2R,3R)-1-carboxy-3,5,7-trihydroxy-4-oxo-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2 H-chromenium | -116.429 | -76.8284 | 39.3642 | -0.236165 |
| 2 | carboxy[(2R,3R)-3,7-dihydroxy-4-oxo-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-5-yl]oxonium | -114.941 | -82.0331 | -31.668 | -1.23969 |
| 3 | carboxy{2,3-dihydroxy-5-[(2R,3R)-3,5,7-trihydroxy-4-oxo-3,4-dihydro-2H-chromen-2-yl]phenyl}oxonium | -102.961 | -72.5893 | 28.4192 | -1.95222 |
| 4 | carboxy{2,3-dihydroxy-5-[(2R,3R)-3,5,7-trihydroxy-4-oxo-3,4-dihydro-2H-chromen-2-yl]phenyl}oxonium | -112.128 | -60.7134 | 47.9748 | -3.43962 |
| 5 | (Z)-sulfo[(2R,3R)-3,5,7-trihydroxy-2-(3,4,5-trihydroxyphenyl)-2,3-dihydro-4H-chromen-4-ylidene]oxonium | -121.908 | -81.9742 | -36.262 | -3.67148 |
Results
The 3D structure of leucotoxin and Cdt is shown in [Table/Fig-1,2]. FASTA sequence of leucotoxin is shown in [Table/Fig-3]. It is made up of 3072 and 4526 amino acids respectively. Their 3D structure is viewed as PDB file with Rasmol structure colour scheme. Alpha helices are shown in magenta, beta sheets in yellow, turns in pale blue, and all other residues in white.
A total of 500 ligands were prepared based on the structure of leucotoxin and Cdt using ACD chemsketch. It was converted to pdb format using OPEN BABEL software. All 500 ligands were then subjected to virtual rapid screening with iGEMDOCK software and six compounds were found to have good fit with a low binding energy. The structure and the IUPAC name of the six ligands were shown in the [Table/Fig-6]. The selected six ligands were then studied for its drug relevant properties.
[Table/Fig-7] depicts the values related to the Lipinski’s rule of Five. From the table it is evident that selected ligand obey the rule. [Table/Fig-8] shows the drug relevant properties of the ligand. It possesses good drug score and drug likeness properties.
Protein-compound interaction profiles of ligands
| Ligand | Molwt | XlogP | Hdonor | Hbond acceptor |
|---|
| carboxy[(2R,3R)-3,7-dihydroxy-4-oxo-2-(3,4,5-trihydroxyphenyl)-3,4-dihydro-2H-chromen-5-yl]oxonium | 397.09 | 2.34 | 5 | 8 |
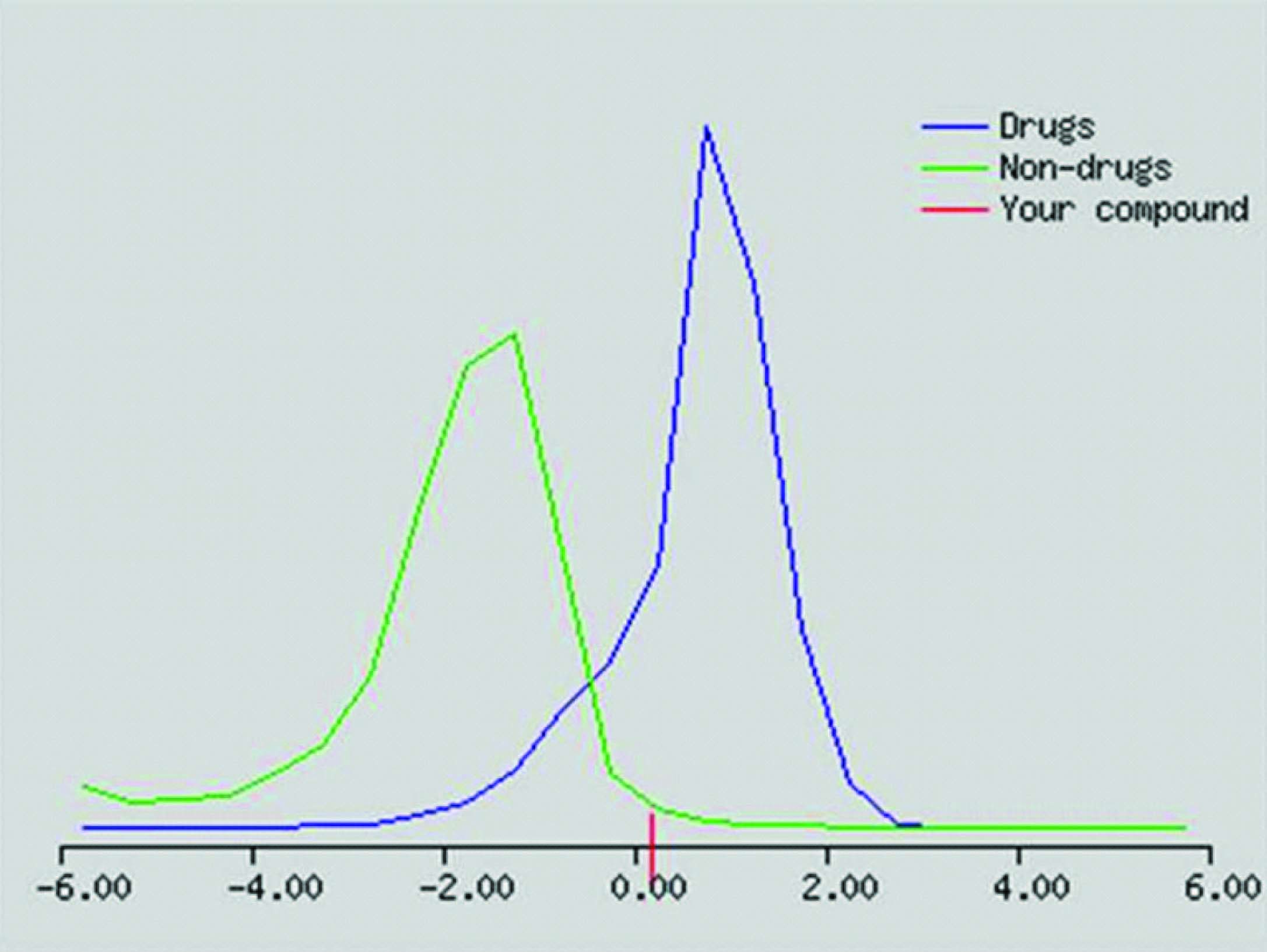
After the confirmation of ADME properties, the six ligands were then subjected to further molecular docking with iGEMDOCK subjecting to accurate docking (very slow docking) by setting population size of 800 is set with 80 generation and 10 solutions. The results were projected in the [Table/Fig-7]. Lower the binding energy, better the fit of ligands. From the table, it is clear the first ligand carboxyl {(2R,3R)-3,7 dihydroxy 4-oxo-2(3,4,5-trihydroxyphenyl)-3,4-dihydro2H-chromen-5-yl} oxonium is found to have excellent fitting compared to other ligands based on the docking energy values [Table/Fig-9] The results clearly indicate that the ligand has a good inhibitory property for leucotoxin and Cdt. Their docking pose was shown in the [Table/Fig-3]. Further this compound has drug likeliness score of 2.01 and a drug score of 0.92 and hence can be a potential drug candidate in the prevention and treatment of aggressive peridontitis caused by Aa.

Discussion
FASTA is a DNA and protein sequence alignment software package first described as FASTP by David J Lipman and William R Pearson in 1985. Before deriving the 3d model of the protein it is necessary for us to know the amino acid sequence of that compound. Since most of the molecules amino acid sequence is available, it is easy to obtain these FASTA sequence. Using this FASTA sequence 3d model of the protein is obtained or derived.
Homology modeling is the easiest way to obtain the 3d model of the protein. It is based on two major observations. The structure of a protein is uniquely determined by its amino acid sequence. During evolution, the structure is more stable and changes much slower than the associated sequence, so that similar sequences adopt practically identical structures, and distantly related sequences still fold into similar structures [11].
This relationship was first identified by Chothia and Lesk (1986) and Sander and Schneider (1991). Protein Data Bank (PDB), Rost (1999) could help in derivation of the 3d structure. As long as the length of two sequences and the percentage of identical residues fall in the region marked as “safe,” the two sequences are practically guaranteed to adopt a similar structure. In the present study 3d structure of leukotoxin was not available. Hence, hemolysin of E-coli was obtained that has the similar amino acid sequence. 3d structure of hemolysin of E-coli was obtained and using homology modelling 3d structure of leukotoxin was derived.
ACD/ChemSketch is a chemical drawing software package from Advanced Chemistry Development, Inc. designed to be used alone or integrated with other applications. Chem Sketch is used to draw chemical structures, reactions and schematic diagrams [9]. It can also be used to design chemistry-related reports and presentations. In the present study we have derived 500 ligands of flavanol.
Several online servers are available to predict ligand binding sites. They are Pocket detection, energy binding site detection, Phylogenetic analysis, binding site database and functional site comparision. Q-SiteFinder is an energy binding site detection online tool. This method analyses interaction energies of a methyl probe with a protein using software developed by Jackson. Probes with favourable interaction energies are retained and clusters of these probes are ranked according to their total interaction energies. The energetically most favourable cluster is then ranked first. Several studies have been conducted using Q-SiteFinder revealed a success rate of 88%-90% in predicting ligand binding site [8,12] Present study utilized this online tool to detect the energy binding site.
Molecular docking is a study of how two or more molecular structures, for example drug and enzyme or receptor of a protein interact with each other. Molecular docking softwares are mainly used in drug research industry. The most important application of docking software is virtual screening (VS). Many tools like GEMDOCK, DOCK, AutoDock, and GOLD have been developed for virtual screening. VS procedure consists of four main steps: preparations of the target protein and the compound library, docking and post-screening analysis. iGEMDOCK is an variation of GEMDOCK which derives the pharmacological interactions from screening compounds without using a set of known active compounds [10,13]. Present analysis uses this iGEMDOCK for docking 500 ligands of flavanol and the toxins of Aa.
The fastest method for evaluating the drug-like properties of a compound is to apply "rule of 5" (RO5) that characterize the molecule. These rules are a set of property values that were derived from classifying the key physicochemical properties of drug compounds. Drug-like molecules, according to Dr. Lipinski, refers to compounds that have sufficiently acceptable ADME properties (absorbtion, distribution, metabolism and excretion) and sufficiently acceptable toxicity properties to survive through the completion of human Phase I clinical trial. Yet, the rule of 5 only underlines properties that would make a compound a likely orally active drug in humans, but clearly these rules do not investigate directly metabolism, probe if a molecule is a frequent hitter or if it contains reactive functional groups. Rule of five includes molecular weight less than or equal to 500, ClogP less than or equal to 5, H- bond donor less than or equal to 5 and H- bond acceptor less than or equal to 10 [14,15] In the present study, obtained drug carboxyl {(2R,3R)-3,7 dihydroxy 4-oxo-2(3,4,5-trihydroxyphenyl)-3,4-dihydro2H-chromen-5-yl} oxonium fulfil all the property of Lipinski rule of five.
Conclusion
The leucotoxin and cytolethal distending toxin of Aa is found to be the major virulence factor involved in the causation of aggressive peridontitis. Hence, the inhibitors of these two proteins can be an effective drug in the prevention and treatment of aggressive peridontitis. In the present study, the ligands were generated and were studied for its ability to inhibit the toxins by molecular docking method. Six ligands with good inhibitory properties were generated among which carboxyl {(2R,3R)-3,7 dihydroxy 4-oxo-2(3,4,5-trihydroxyphenyl)-3,4-dihydro2H-chromen-5-yl} oxonium, a novel compound is found to be excellent drug candidate based on the molecular docking studies and its ADME properties.
[1]. Zambon JJ, Actinobacillus actinomycetemcomitans in human periodontal diseaseJ Clin Periodontol 1985 12:1-20. [Google Scholar]
[2]. Kler S, Malik R, An Update on the Virulence Factors of Actinobacillus actinomycetemcomitans - A Systematic Review. Research & ReviewsA Journal of Dentistry 2010 1:1-10. [Google Scholar]
[3]. Henderson B, Nair SP, Ward JM, Wilson M, Molecular pathogenicity of the oral opportunistic pathogen Actinobacillus actinomycetemcomitansAnnu Rev Microbiol 2003 57:29-55. [Google Scholar]
[4]. Haubek D, Ennibi OK, Poulsen K, Benzarti N, Baelum V, The Highly Leukotoxic JP2 Clone of Actinobacillus actinomycetemcomitans and Progression of Periodontal Attachment LossJ Dent Res 2004 83:767-70. [Google Scholar]
[5]. Lally ET, Hill RB, Kieba IR, Korostoff J, The interaction between RTX toxins and target cellsTrends Microbiol 1999 7:356-61. [Google Scholar]
[6]. Thelestam M, Frisan T, Cytolethal distending toxinsRev Physiol Biochem Pharmacol 2004 152:111-33. [Google Scholar]
[7]. Mayer MP, Bueno LC, Hansen EJ, Di Rienzo JM, Identification of a cytolethal distending toxin gene locus and features of a virulence-associated region in Actinobacillus actinomycetemcomitansInfect Immun 1999 67:1227-37. [Google Scholar]
[8]. Laurie ATR, Jackson RM, Q-Site Finder: an energy-based method for the prediction of protein-ligand binding sitesBioinformatics 2005 21:1908-16. [Google Scholar]
[9]. ACD/ChemSketch Freeware, version 10.00, Advanced Chemistry Development, Inc., Toronto, ON, Canada, 2012 [Google Scholar]
[10]. Yang JM, Chen CC, "GEMDOCK: A generic evolutionary method for molecular Docking."Proteins: Structure, Function, and Bioinformatics 2004 55:288-304. [Google Scholar]
[11]. Krieger E, Nabuurs SB, Vriend G, HOMOLOGY MODELING. In: Bourne PE, Weissig H, editorStructural Bioinformatics 2003 Wiley-Liss, Inc:507-21. [Google Scholar]
[12]. Laurie ATR, Jackson RM, Methods for the Prediction of Protein-Ligand Binding Sites for Structure- Based Drug Design and Virtual Ligand ScreeningCurrent Protein and Peptide Science 2006 7:395-406. [Google Scholar]
[13]. Hsu KC, Chen YF, Lin SR, Yang JM, iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysisBioinformatics 2011 12(1):S33 [Google Scholar]
[14]. Lipinski CA, Drug-like properties and the causes of poor solubility and poor permeabilityJournal of Pharmacological and Toxicological Methods 2000 44:235-49. [Google Scholar]
[15]. Keller TH, Pichota A, Yin Z, A practical view of ‘druggability’Current Opinion in Chemical Biology 2006 10:357-61. [Google Scholar]