Primary MPNST in Childhood- A Rare Case Report
Sandip Kudesia1, Aparna Bhardwaj2, Brijesh Thakur3, Sanjeev Kishore4, Neelima Bahal5
1 Professor & Head, Department of Pathology, SGRRIM & HS, Dehradun, Uttrakhand, India.
2 Associate Professor, Department of Pathology, SGRRIM & HS, Dehradun, Uttrakhand, India.
3 Assistant Professor, Department of Pathology, SGRRIM & HS, Dehradun, Uttrakhand, India.
4 Professor, Department of Pathology, SGRRIM & HS, Dehradun, Uttrakhand, India.
5 Juniour resident II, Department of Pathology, SGRRIM & HS, Dehradun, Uttrakhand, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Brijesh Thakur, Assistant Professor, Department of Pathology, SGRRIM & HS, Mahant Indresh Hospital, Patel Nagar, Dehradun-248001, Uttrakhand, India. E-mail : drbrijeshthakur03@gmail.com
Malignant peripheral nerve sheath tumour usually occurs between 20-50 years of age, comprising about 5-10% of soft tissue sarcomas. Only 1.7% of them have been reported to occur in children < 5 months of age according to the literature. Here, we are describing 18 mnth old male child presented with a swelling in the lower back. MRI showed a sacrcoccygeal swelling extending to and communicating with CSF at lower sacral level. Birth history of the child was normal with normal apgar score. The histological diagnosis was malignant peripheral nerve sheath tumour. IHC showed focal positivity of GFAP and S100. Primary spinal MPNST in children are rarer. A careful neurological examination is warranted in children. Early diagnosis and referral to multidisciplinary team are important in ensuring the best diagnosis and optimal therapy in this young age.
Child, Lipomeningocele, Neurofibromatosis, Neural, Sacral
Case Report
An 18-months-old male child was referred to the neurosurgery OPD with a swelling in the lower back and weakness of both the lower limbs. The swelling was initially noticed by his mother at 4 months of age and was increasing gradually in size since then. There was no history of trauma, urinary or fecal complaints. Antenatal ultrasound reports had not reported any congenital abnormality. The swelling was present in midline of the lower back, extending to left paramedian region 5 cm cranial to anal orifice, with normal overlying skin.
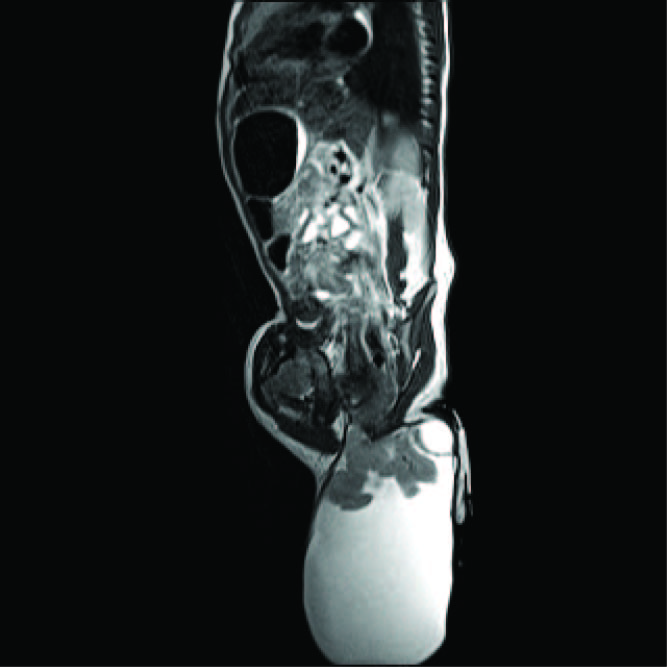
Lumbosacral MRI examination showed a sacrococcygeal lesion extending and communicating with the CSF space at lower sacral level to the subcutaneous tissue with flaring of lamina at S3 level and outpouching in lower back [Table/Fig-1]. There was no cord teethering. Based on these findings a diagnosis of sacral lipomeningocele was made. Intraoperative findings showed a sacrococcygeal swelling measuring 9 x 5 x 2.5 cm. present in subcutaneous plane and extending deeply upto dura.
MRI film showing a sacrococcygeal swelling,
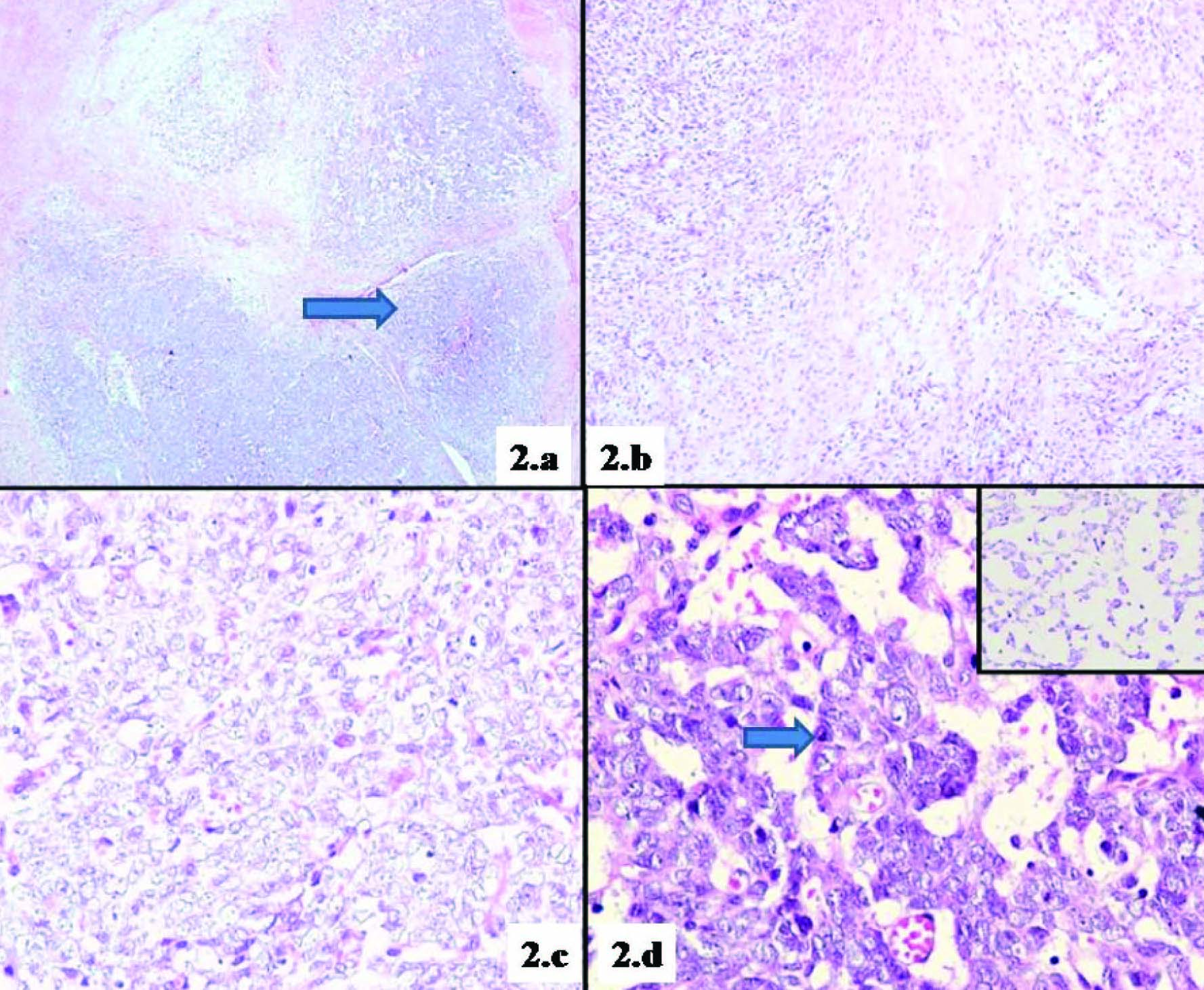
On gross examination, excised tumour was creamy white in colour and outer surface was nodular. Microscopic examination of the tumour was performed. It showed a cellular tumour exhibiting a vague nodular pattern composed of highly compact cellular zones amidst which were interspersed loose myxoid areas [Table/Fig-2a]. The cellular area showed fascicles of spindle cells with areas of randomly and loosely arranged Schwann cells alternating with less cellular areas creating a marbelized appearance [Table/Fig-2b]. Here the tumour cells revealed sheets of large polygonal cells with enlarged nucleus exhibiting irregular margins a few of which revealed prominent nucleoli and scant cytoplasm. Focal areas show myxoid material intervening with these neoplastic cells [Table/Fig-2c,d]. Mitotic figures were numerous (4-5/10 HPF) with occasional atypical mitosis. Foci of necrosis, normal appearing nerve fibres and normal adipose tissue were also observed. The most remarkable features were the presence of numerous thin walled congested blood vessels amidst tumour.
a. Tumour cells showing a nodular growth pattern (H&E, 40X), b. Neoplastic cells showing a marbelized appearance created by alternating dense to less cellular areas (H&E,100X), c. Neoplastic cells showing solid areas (H&E, 400X), d. Neoplastic cells in myxoid background (H&E, 400X), Inset showing neoplastic cells floating in myxoid areas (H&E, 400X),
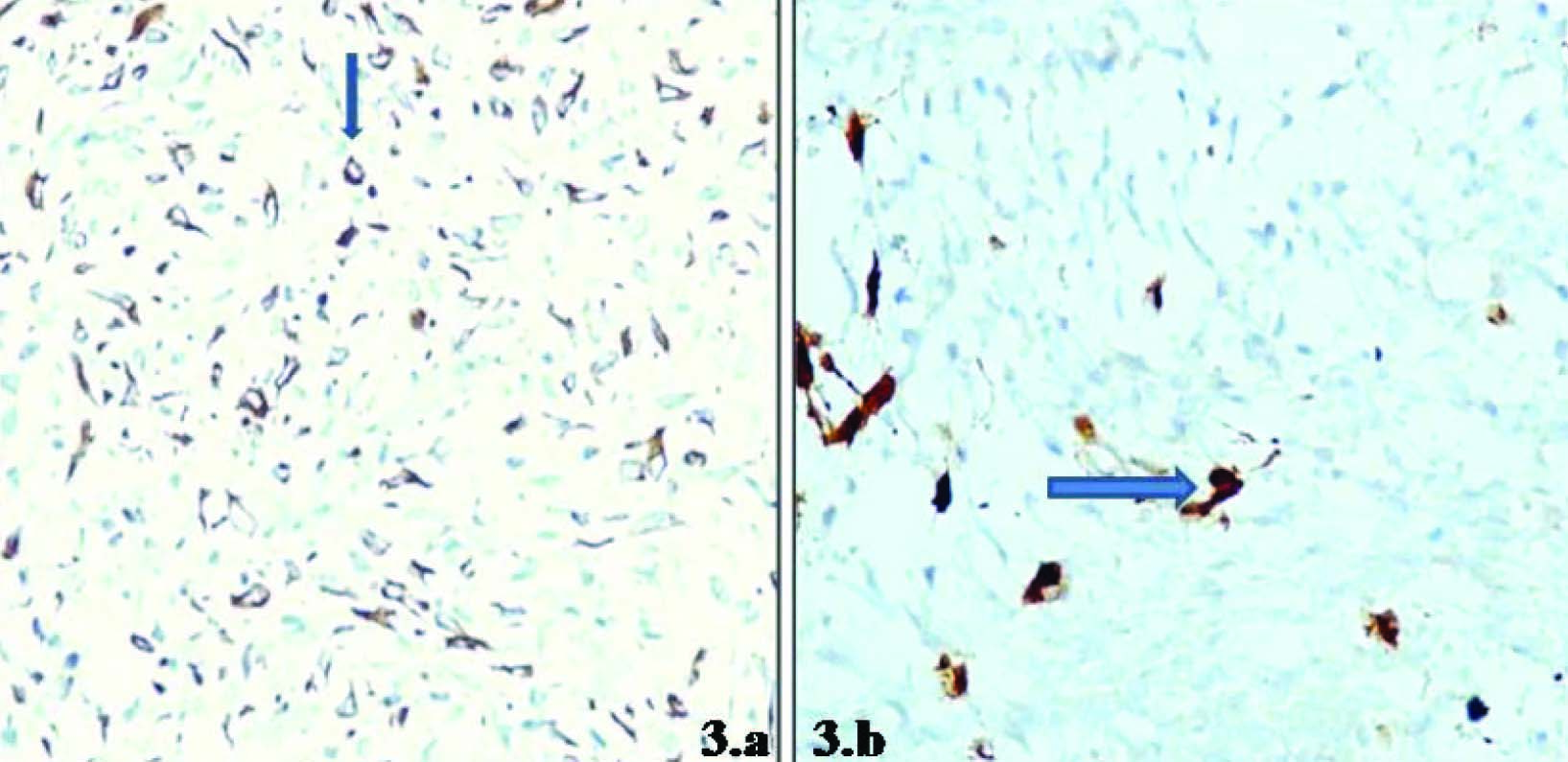
Immunohistochemistry (IHC) was done employing GFAP and S100 that showed focal positivity [Table/Fig-3a,b]. Based on these pathological and immunohistological findings, the tumour was diagnosed as malignant peripheral nerve sheath tumor (MPNST).
a. Focal and discrete GFAP positivity, b. Focal S-100 positivity
Discussion
MPNST, originating from major nerve trunks such as brachial plexus, sacral plexus or sciatic nerve or from cells associated with nerve sheaths as Schwann cell, perineural cells or fibroblasts are rare and their occurrence in children is rarer still [1–3]. Only 1.7% of the cases have been reported to occur in children less than five year of age. Depending on its location and amount of nerve involvement, MPNST can present as a painful or painless mass. A sarcoma is defined as a MPNST when at least one of the following criteria is met; origin from peripheral nerve; origin from pre-existing nerve sheath tumour (neurofibroma) and / or demonstration of Schwann cell differentiation on histologic examination. MPNSTs located in the trunk and extremities are usually high grade and clinically aggressive with poor prognosis [4,5].
An association between MPNST and NF–1 has been well documented compared with patients who do not have neurofibromatosis (NF–1). Patient with NF–1 are younger in age and have a central distribution of primary tumour [1,6]. The incidence of primary MPNSTs of the spinal canal are extremely rare and are associated with poor clinical outcomes [7].
Delay in diagnosis of this soft tissue tumour is common. Patients with intraspinal tumours may be erroneously diagnosed initially as poliomyelitis, spina bifida, cerebral palsy, muscular dystrophy and Gullian – Barre syndrome [8]. Radiographic imaging and electrophysiologic studies are helpful in the work-up of MPNST cases, though findings do not allow differentiation from other tumours. Hence, histopathological diagnosis of the tissue is necessary. The gross pathology of these tumours is quite distinctive in classical form. It grossly shows a large fusiform, eccentric mass in a major nerve. Most of these tumours are deeply situated and exhibit a mean diameter of 5 cm or more. Their appearance is fleshy, opaque surface with areas of secondary haemorrhage and necrosis [1].
Microscopic and immunohistochemical analysis are imperative to establish a correct diagnosis. The histologic features of MPNSTs show considerable variation. Several features that favours the histological diagnosis includes perivascular hypercellularity, hyperchromatic wavy nuclei, high mitotic activity, necrosis and focal S-100 positivity [9]. Distinguishing Schwannoma from MPNST is important because schwanoma has a benign course with rare malignant degeneration. Cellular Schwannomas although exhibit brisk mitosis, can be identified by features such as Antoni B areas, secondary degenerative change and hyalinised thick walled blood vessels. Conventional MPNST show a geographic type of necrosis, anaplastic cells and widespread mitosis (usually > 10/10HPF) [10]. Immunohistochemistry is employed for identifying nerve sheath differentiation. Nerve sheath tumours usually display positivity for S-100 antigen, leu – 7, mylein basic protein and PG P 9.5 [11].
Cellular Schwannoma is a close differential diagnosis of MPNST. Immunoreactivity employing antibodies to S-100 is the key differentiating point between these two entities. A strong S-100 positivity is observed in cellular Schwannoma where as focal positivity is observed in MPNST. Reactivity for S-100 protein may be variable in MPNST because of differentiation and poor dedifferentiation.
Younger age, higher grade, stage and tumour size significantly relate to aggressiveness of disease. As of now, surgery with wide excision with options of adjuvant chemotherapy and/or radiotherapy is the treatment of choice for a better outcome [6].
Conclusion
Historically MPNST have been difficult to diagnose and treat. There is usually a delay in the diagnosis of this soft tissue tumour and patients with intraspinal tumours can have initial erroneous diagnosis because of variable or no neurological symptoms. A careful examination is warranted in children as early diagnosis and reference is important in ensuring the optimal treatment modality at this young age.
[1]. Enzinger FM, Malignant tumours of peripheral nerves. In Weiss SW, Goldblum JR, edsEnzinger and Weiss’s Soft Tissue Tumours 2001 4th edSt. Louis, MOMosby:1209-40. [Google Scholar]
[2]. Kourea HP, Bilky MH, Leung DHY, Lewis JJ, Woodruff JM, Subdiaphgramatic and intrathoracic paraspinal malignant peripheral nerve sheath tumours. A clinicopathologic study of 25 patients and 26 tumoursCancer 1998 82:2191-203. [Google Scholar]
[3]. Acharya R, Bhalla S, Sehgal AD, Malignant peripheral nerve sheath tumour of the cauda equineNeurol Sci 2001 22:267-70. [Google Scholar]
[4]. Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HH, Ilstup H, Malignant peripheral nerve sheath tumours. A clinicopathologic study of 120 casesCancer 1986 57:2006-21. [Google Scholar]
[5]. Lewis JJ, Brenna MF, Soft tissue sarcomasCurr Probl Surg 1996 33:817-80. [Google Scholar]
[6]. Rekhi B, Ingle A, Kumar R, De Souza MA, Dikshit R, Jambhekhar NA, Malignant peripheral nerve sheath tumours: Clinicopathological profile of 63 cases diagnosed at tertiary cancer referral center in Mumbai, IndiaIndian J Pathol Microbiol 2010 53(4):611-18. [Google Scholar]
[7]. Seppala MT, Hallia MJJ, Spinal malignant nerve sheath tumour or cellular Schwannoma? A stricking difference in prognosisJ Neurosurg 1993 79:528-32. [Google Scholar]
[8]. Ogawa BK, Skaggs DL, Kay RM, Malignant peripheral nerve sheath tumor of Lumber spineThe American Journal of Orthopedics 2009 38(5):89-92. [Google Scholar]
[9]. Evans DG, Baser ME, McGaurharn J, Sharif S, Howard E, Moran A, Malignant peripheral nerve sheath tumours in neurofibromatosisJ Med Genet 2002 39:311-14. [Google Scholar]
[10]. White W, Shuii MH, Rosenblum MK, Erlandson RA, Woodruff JM, Cellular Schwannoma. A clinicopathologic study of 57 patient and 58 tumoursCancer 1990 66:1266-75. [Google Scholar]
[11]. Celli P, Cervoni L, Taratino R, Fortuna A, Primary spinal malignant Schwannomas: clinical and prognostic remarkesActa Neurochin (Wien) 1995 135(1-2):52-55. [Google Scholar]