Haemoglobin S Interaction with Beta Thalassaemia- A Case Report from Assam, India
Mauchumi Saikia Pathak1, Monalisha Saikia Borah2, Dulal Kalita3
1 Professor, Department of Biochemistry, Gauhati Medical College & Hospital, Guwahati, Assam, India.
2 Senior Research Fellow, Department of Biochemistry, Gauhati Medical College & Hospital, Guwahati, Assam, India.
3 Associate Professor, Department of Paediatrics, Gauhati Medical College & Hospital, Guwahati, Assam, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Mauchumi Saikia Pathak, Professor, Department of Biochemistry, Gauhati Medical College & Hospital, Narkasur Hill top, Guwahati-781032, Assam, India.
Phone: 9435733878, E-mail : mauchumip@gmail.com
Interaction of Hb S with beta thalassaemia is being reported here as this type of case is rare. Hb S (β6 glu→val) is a genetic disorder which occurs due to beta globin gene mutation of haemoglobin. In India, the Hb S is prevalent in the central part, in the eastern, western and southern tribal belt regions and among the tea tribe communities of Assam. The Hb S carriers (Sickle cell trait) leads a normal life but the Sickle cell disease patients show certain clinical manifestation like joint pain, anaemia and jaundice. The HPLC report of the patient showed Compound heterozygous for Hb S- β thalassaemia. The complete blood count was measured in automated haematology analyser. Mutational pattern of the beta thalassaemia as well as the presence of Hb S gene was detected by PCR. The case showed severe clinical manifestations and transfusion was required due to inheritance of the IVS 1-5 G →C β- thalassaemia mutation with the Hb S gene.
ARMS-PCR, Compound heterozygous, Hb S- β thalassaemia, HPLC, Sickle cell
Case Report
A 3-year-old female patient was referred from the Department of Paediatrics, Gauhati Medical College & Hospital, Assam, India for Haemoglobin typing. The patient was severely anaemic and complained for weakness, joint pain and stomach pain. The parents of the child were asymptomatic but both complained for weakness. The patient and both the parents were from Tea tribe community.
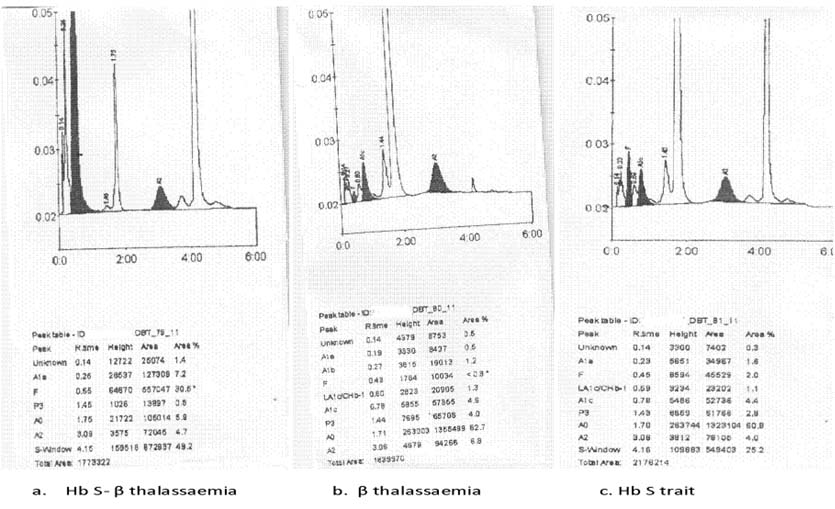
Blood samples of patient along with parents’ were collected in EDTA coated vaccutainers after obtaining written informed consent. The necessary information was recorded in proforma. The complete blood count was measured using the automated haematolgy analyser (pocH100i, SYSMEX) and the Haemoglobin pattern was detected by the HPLC based Haemoglobin Typing System (D10, BIORAD). The patient was severely anaemic with 3.4 g/dl Haemoglobin. The haematological data of the family are shown in [Table/Fig-1]. The HPLC report showed that the patient was Compound heterozygous for Hb S- β thalassaemia, the father had β- thalassaemia trait, and the mothers’ chromatogram revealed Sickle cell trait [Table/Fig-2] (Chromatogram of the family). Further confirmation of diagnosis was done by Molecular study. DNA extraction was done by column based DNA extraction kit and then ARMS- PCR was done to find out the beta thalassaemia mutational pattern and to confirm the Hb S mutation. The patient and her father were beta thalassaemia carrier and were detected with IVS 1-5 G→C mutation [Table/Fig-3] (Gel picture showing IVS 1-5 G →C mutation). Hb S mutation was confirmed by doing PCR and both the patient and her mother showed positive for the Hb S mutation and were carrier of the Hb S gene [Table/Fig-4] (Gel picture showing Hb S mutation).
The haematological data of the family
| Parameters | Patient | Father | Mother |
|---|
| WBC x 103 ul | 16.4 | 4.8 | 10.8 |
| RBC 106 ul | 1.17 | 3.0 | 4.6 |
| HGB g/dl | 3.4 | 9.1 | 11.8 |
| HCT % | 10.7 | 247.3 | 35.2 |
| MCV f1 | 91.5 | 88.6 | 75.2 |
| MCH pg | 29.1 | 29.5 | 24.8 |
| MCHC g/dl | 31.8 | 33.3 | 33.0 |
Chromatograms of the family
Gel picture showing IVS 1-5 G C mutation

Gel picture showing Hb S mutation

Discussion
Haemoglobinopathies occurs due to structural defect in the globin gene, whereas the thalassaemias are due to defect in the production of globin chain. The sickle haemoglobin (Hb S) is of clinical importance because of their worldwide prevalence. Sickle cell anaemia (Hb SS) is the most common heritable haematological disease affecting humans [1]. It is seen frequently in Africa and amongst black populations in North and South America and the West Indies. It is also found in certain regions in Greece, Turkey, Italy, Middle East and India [2]. In some parts of Africa, around 45% of the population have sickle cell trait and about 8% of blacks in United States, Latin America and Caribbean carry the sickle cell gene [1]. In North- East India, Sickle cell haemoglobin (Hb S, β6 glu→val) is mainly prevalent in the Tea garden labour communities of Assam [3]. The average frequency of Sickle cell disorders in India is 4.3%. The highest prevalence has been recorded in the state of Orissa (1-44.4%), followed by Madhya Pradesh (1-40.0%; including Chhattisgarh), Tamil Nadu (1-40.0%), Andhra Pradesh (1-35.7%), Assam (1-35.5%), Maharashtra (0.8-35.0%), Gujarat (1-31.4%), Kerala (1-30.0%), Uttar Pradesh (1.5-18.5%), Karnataka (1-8.0%) [4]. A case of Compound heterozygous for Hb S- β thalassaemia was previously reported with severe anaemia, massive hepatomegaly, splenomegaly and acute Chest syndrome [5]. In our case also the patient had severe anaemia, weakness and complained of joint pain and stomach pain. On physical examination hepatosplenomegally was observed in the patient. Only after our diagnosis report, the parents of the patient became aware about their Hb variant carrier state.
Conclusion
The association of beta thalassemia with Hb S is rare, which was reported as 1.7% [6]. So this case of interaction of Hb S with beta thalassaemia is being reported here as this type of case is rarely encountered.
[1]. Wang WC, Sickle cell anaemia and other Sickling Syndromes. In: Greer JP, Foester J, Rodgers GM, Paraskevas, Glader B, Arber DA, Means RT, Jr, editorsWintrobe’s Clinical Hematology 2009 12th edLippincott William & Wilkins:1038-82. [Google Scholar]
[2]. Das Reena, Disorders of Haemoglobin Structure and Synthesis. In: Saxena R, Pati HR, Mahapatra M, editorsde Gruchy’s Clinical Haematology in Medical Practice 2013 6th edWiley India Pvt. Ltd:120-45. [Google Scholar]
[3]. Balgir RS, Sharma SK, Distribution of sickle cell haemoglobin in IndiaIndian Journal of Hematology and Blood Transfusion 1988 6:1-14. [Google Scholar]
[4]. Balgir RS, Epidemiology, Population health genetics and phenotypic diversity of Sickle cell disease in IndiaThe Internet Journal of Biological Anthropology 2007 1(2):1939-4594. [Google Scholar]
[5]. Jailkhani R, Patil VS, Kulkarni SP, Pervatikar S, Jayashankara BB, An interesting case of Compound heterozygous for Sickle cell- β thalassaemia presenting with acute chest syndromeJ Clin and Diagn Res 2010 4:2291-96. [Google Scholar]
[6]. Balgir RS, Spectrum of Hemoglobinopathies in the state of Orissa, India: A ten years cohort studyJAPI 2005 53:1021-06. [Google Scholar]