Haematological Characterisation and Molecular Basis of Asian Indian Inversion Deletions Delta Beta Thalassemia: A Case Report
Jitender Mohan Khunger1, Monika Gupta2, Rekha Singh3, Rohit Kapoor4, Hare Ram Pandey5
1 Consultant Haematologist & Associate Professor, Department of Haematology, Vardhman Mahavir Medical College and Safdar Jang Hospital, New Delhi, India.
2 Pool Officer, Department of Haematology, All India Institute of Medical Sciences, New Delhi, India.
3 Research Associate, Department of Haematology, All India Institute of Medical Sciences, New Delhi, India.
4 Medical Officer, CGHS Dispensary, Krishna Nagar, Delhi, India.
5 Research Associate,Department of Haematology, All India Institute of Medical Sciences, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr.Jitender Mohan Khunger, First Floor, Department of Haematology, VMMC & Safdarjang Hospital, New Delhi-110029, India. Phone : +91-9810288232, Email : drjmkhunger@rediffmail.com, drjmkhunger@gmail.com
The hereditary persistence of fetal hemoglobin (HPFH) and delta beta thalassemia are heterogeneous disorders characterised by increased levels of fetal hemoglobin and high level of this Hb continues in adulthood. The distinction between these two conditions is not always possible with routine hematologic analysis and molecular characterisation of the defect is required. We encountered such a rare case of δ β thalassemia in a 10-year-old male child who presented with features of thalassemia intermedia. Hemoglobin analysis showed 100% HbF while molecular analysis revealed Asian Indian inversion-deletion GγAγ(δ β) zero thalassemia.
Fetal haemoglobin, Genetic counselling, GγAγ(δ β)0 thalassemia, Gap- PCR, Hereditary persistence of fetal hemoglobin, Hematology analyser, Thalassemia intermedia
Case Report
We hereby describe a rare case of δ β thalassemia in a young Indian male child who was diagnosed as homozygous δ β-thalassemia after gene mutation studies.
A young 10-year-old male child presented in outpatient department (OPD) of Hematology at All India Institute of Medical Sciences, New Delhi, with complaints of pallor, weakness and jaundice for the last 5-6 yrs. There was no history of blood transfusions or drug intake. Parents had non consanguineous marriage and with no similar findings in the family.
On examination he had features of thalassemia intermedia phenotype with anaemia, jaundice and mild hepatosplenomegaly. Other systemic examinations were within normal limits. Investigation on automated haematology analyser(Sysmex K-450. Kobe, Japan), showed that he had haemoglobin 9.0 gm%, MCV 78.8 fl, MCH 20.3pg, RBCs count 4.9 ×106/μl, and RDW-CV 16%. Giemsa stained peripheral smear examined for red cell morphology showed microcytic hypochromic red cells. Iron studies were with in normal limits. Quantitative test was performed to determine levels of HbA, HbA2 and HbF by High performance liquid chromatography (HPLC) (Bio-rad-variantTMBioRad, CA, USA). It showed 100% HbF in patient’s blood sample. Provisional diagnosis of hereditary persistence of fetal haemoglobin was considered. Parents’ HPLC was also done, which showed high HbF and normal HbA2. Informed consent was obtained from parents for performing haematological & molecular investigations of the patient and parents. Hematological and molecular details of the patient and parents are mentioned in tabulated form [Table/Fig-1].
Results of the patient and parents are listed in the table
| Parameter | Patient | Female | Single |
|---|
| Sex/age (yrs) | M/10 yrs | F/35 yrs | M/38 yrs |
| Hb (g/dL) | 9.0 | 11.6 | 13.0 |
| Hct (%) | 35.0 | 41.4 | 45.5 |
| MCV (fL) | 78.8 | 83.8 | 87.5 |
| MCH (pg) | 20.3 | 23.5 | 25.0 |
| MCHC (g/dL) | 25.7 | 28.0 | 28.6 |
| Hb typing/HPLC |
| HbA2 % | 0.0 | 2.5 | 2.5 |
| HbF % | 100 | 16.6 | 18.0 |
| Mutation study for δ β- thalassemia | Homozygous for both breakpoint A and B of the Asian-Indian Inv. deletion | Compound heterozygous for breakpoint A and B of the Asian-Indian Inv. deletion | Compound heterozygous for breakpoint A and B of the Asian-Indian Inv. deletion |
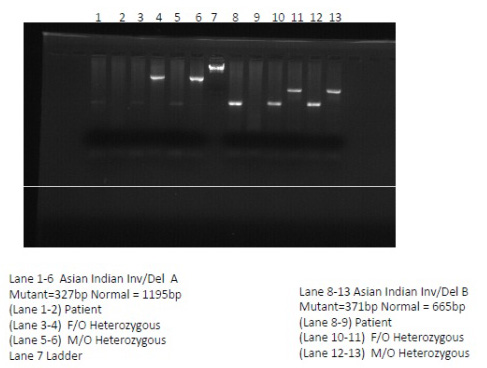
DNA extraction was done from peripheral blood leucocytes by phenol chloroform method. Molecular analysis for beta mutation was done according to published literatures. Asian Indian inversion-deletion GγAγ(δ β)0 thalassemia type A and type B mutations were identified by Gap-PCR. The amplicons were electrophoresed on 1.2% agarose gel containing ethidium bromide and photographed under UV light through Image Capture System L-PIX Touch (Loccus Biotechnology). The size of the fragments obtained after amplification for the mutations analysed are shown in [Table/Fig-2]. This inversion-deletion is characterised by a 5’ deletion of 11.5kb (deletion A), and a 3’ deletion of 1.6 kb (deletion B) flanking an inverted region of DNA of 7.6 kb.
PCR done showing fragments obtained after amplication for Mutation
Discussion
Delta beta GγAγ(δ β)0 thalassemia results from deletion of both δ and δ genes but with preservation of the γ genes. As homozygotes have no δ or β genes, they cannot synthesise HbA2/A. HbF comprises 100% of hemoglobin. Heterozygotes for δ β-thalassemia tend to have a modest elevation of HbF (5-20%) with hypochromic microcytic red cell indices [1]. Homozygotes are not anaemic as fetal hemoglobin has a higher oxygen affinity than HbA, hence patients who have β+ or β0 mutation may have thalassemia intermedia rather than thalassemia major [2]. HPFH is associated either with the occurrence of single-point mutations in the promoter region of two γ chains or β-globin gene cluster deletions. It is usually characterised in heterozygotes by higher levels of HbF (upto 30%) with normal red cell indices [3]. Individuals with these disorders exhibit milder clinical symptoms than those with typical β-thalassemia, due to the beneficial effect of HbF on red blood cell production and survival [4,5]. There is a thin line of clinical and haematological difference between HPFH and δ β-thalassemia. Therefore, the level of HbF alone sometimes cannot differentiate the two conditions and DNA analysis of the molecular defect is required.
In this case report, the patient had a thalassemia intermedia phenotype with significant microcytic hypochromic red cells. His Hb analysis revealed 100% HbF. The information obtained from PCR study aided in the rapid diagnosis and showed a homozygous δ β thalassemia of the Asian Indian inversion-deletion in the patient. Both his parents showed a compound heterozygous for δ β thalassemia of the Asian Indian inversion-deletion. As per the literature, the complex double deletion inversion rearrangement GγAγ(δ β)0 appears to be a common form of δ β thalassemia found in Central Asia, India, Kuwait and Iran, while, GγAγ(δ β)0 thalassemia and HPFH-6 caused by DNA deletions are the most common high Hb F determinants in Thailand [6,7]. Since the high fetal hemoglobin determinants are found in Southeast Asian countries, compound heterozygotes of these conditions with β thalassemia or other haemoglobinopathies are not uncommon [7].
Conclusion
Differential diagnosis of these conditions is therefore important for providing an appropriate treatment and genetic counselling to the patient. Identification of these abnormalities also facilitate in prevention and control program of thalassemia as well as in prenatal and newborn screening for hemoglobinopathies in the region. The characterisation methods used were necessary along with routine hemoglobin analysis to determine the genotype properly as diagnosis requires both family study and DNA analysis.
[1]. Supan F, Yutthana P, Satja S, Goonnapa F, Kanokwan S, Molecular and hematological characterization of hereditary persistence of fetal hemoglobin-6/ Indian deletion –inversion Gγ(Aγ δ β)0-thalassemia and Gγ(Aγ δ β)0-thalassemia/ HbE in Thai patientsAmerican Journal of Hematology 2002 71:109-13. [Google Scholar]
[2]. Barbara BJ, The alpha, beta, delta and gamma thalassemias and related conditions. In : Bain BJ edHemoglobinopathy diagnosis 2006 second editionBlackwell publishing:63-138. [Google Scholar]
[3]. Carrocini GCS, Ondei LS, Zamaro PJA, Bonini-Domingos CR, Evaluation of HPFH and δ β- thalassemia mutations in a Brazilian group with high Hb F levelsMol. Res 2011 10(4):3213-19. [Google Scholar]
[4]. Rochette J, Craig JE, Thein SL, Fetal Haemoglobin levels in adultsBlood Rev 1994 8:213-24. [Google Scholar]
[5]. Wood WG, Increased fetal hemoglobin in adult lifeBailliere’s Clin Haematol 1993 6:177-213. [Google Scholar]
[6]. Pandey S, Pandey S, Ranjan R, Mishra R, Sharma M, Saxena R, Phenotypic heterogeneity of Asian Indian Inversion Deletion GγAγ( δ β)0 breakpoint A and breakpoint BInd J Clin Biochem 2013 28(1):98-101. [Google Scholar]
[7]. Panyasai S, Fucharoen S, Surapot S, Fucharoen G, Sanchaisuriya K, Molecular basis and hematologic characterization of δ γ- thalassemia and hereditary persistence of fetal haemoglobin in ThialandHaematologica 2004 89:777-81. [Google Scholar]