Overview
The healthy endothelium plays a pivotal role in regulating vascular homeostasis. A plethora of factors converge to generate endothelial dysfunction, which seems to be the main road to atherosclerosis. Our journey down the rabbit hole starts with the well-known gate keeper, nitric oxide (NO); and we will take a look at the struggle between the good (Paraoxonase) and the bad (Homocysteine), to seek dominion over the microworld which is guarded by NO.
Nitric Oxide: A Radical in Charge
NO is a very simple free radical which plays an important regulatory role in many tissues, mainly vascular system. NO is produced from the amino acid L-arginine by the enzymatic action of nitric oxide synthase (NOS). Vasoprotective NO dilates all types of blood vessels by stimulating soluble guanylyl cyclase and increasing cyclic guanosine monophosphate in smooth muscle cells (SMCs). NO, released towards the vascular lumen, is a potent inhibitor of platelet aggregation and adhesion [1,2].
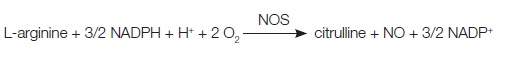
Co-factors for NOS include oxygen, NADPH, tetrahydrobiopterin (BH4) and flavin flavin adenine nucleotides. The synthesis of NO can be inhibited by naturally occurring analogues of the NO precursor, l-arginine, such as asymmetric dimethylarginine (ADMA), whereas dimethylarginine dimethylaminohydrolase 2(DDAHII) is recognized as a protective factor which improves the endothelial function [3,4]. There are two endothelial forms of eNOS: constitutive eNOS (cNOS) and inducible eNOS (iNOS) [5]. The endothelial isoform (eNOS) is the primary signal generator in the control of vascular tone, insulin secretion, airway tone, and it is involved in regulation of cardiac function and angiogenesis. In addition to endothelial eNOS, there is a neural NOS (nNOS) that serves as a transmitter in the brain and in different nerves of the peripheral nervous system [Table/Fig-1].
NO protects against atherosclerotic disease through anti-inflammatory and anti-oxidant effects, and loss of NO bioavailability is a key feature of endothelial dysfunction which precedes the appearance of atherosclerosis [6,7].
Nitric Oxide and Homocysteine
Of concern are diseases and conditions which are associated with malfunction of NO, which apply to hyperhomocysteinaemia (HHcy) [Table/Fig-2]. The question is: how can Hcy modulate NO synthase?
Today, it is believed that the mechanism of atherosclerotic disease in HHcy is directly related to vascular endothelial cell damage which leads to vascular endothelial dysfunction, and enhanced oxidative stress [8-11]. Oxidative radicals which are generated by Hcy inevitably initiate the oxidative degradation of cell membrane lipids of the endothelium, leading to loss of membrane function. Hcy can enhance reactive oxygen species (ROS) production. So, the main molecules which are responsible for decrease in NO levels in HHcy patients are probably ROS [8,12,13].
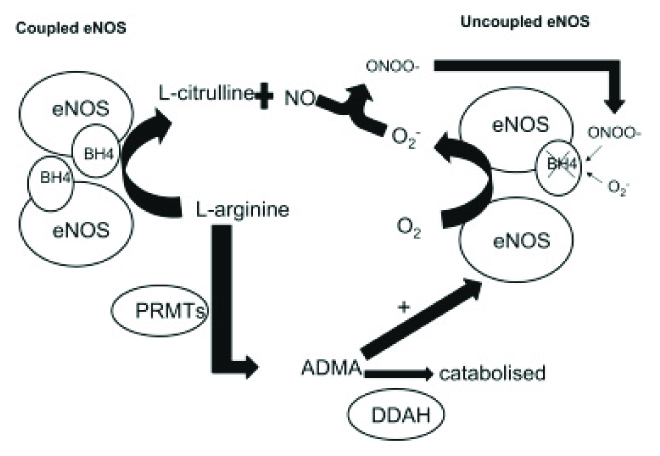
The reduction in BH4 availability, followed by the uncoupling of eNOS, is the significant mark in Hcy-mediated oxidative stress [14,15]. This is an exact feature of endothelial dysfunction that directly precedes the appearance of atherosclerosis [16]. Importantly, BH4 is an essential cofactor for eNOS. When BH4 levels are inadequate, eNOS is no longer coupled with L-arginine oxidation, which results in ROS rather than NO production, thereby inducing vascular endothelial dysfunction [17]. So, the association between Hcy and endothelial dysfunction depends largely on its damaging effect on eNOS coupling. Indeed, He et al., reported that chronic HHcy may induce dysfunction of the coronary artery endothelium through uncoupling of eNOS, as was shown by the low levels of NO and BH4 [Table/Fig-3] [16]. Tawakol et al., indicated that HHcy was associated with impaired endothelium-dependent vasodilation in humans and they suggested that the bioavailability of NO was decreased in HHcy [18] . Stühlinger et al., showed that, another possible method of NOS activity regulation by Hcy was action through direct dimethylarginine dimethylaminohydrolase binding via disulfide bounds and further asymmetric dimethylarginine accumulation, that disrupted NOS activity [19].
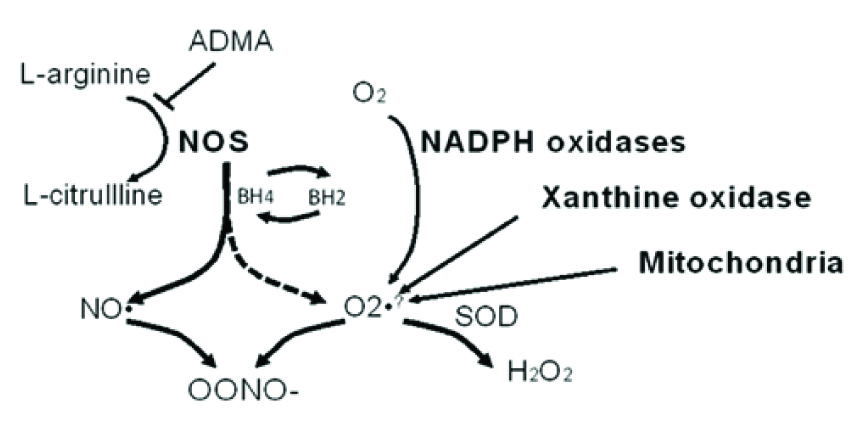
Hcy also induces NADPH oxidase activity, which contributes to increased ROS production. Hcy-induced ROS was presented in HHcy apoE_/_ mice, to upregulate the expression and translocation of redox factor 1 through NADPH oxidase, that in turn accelerated atherosclerosis development [16]. Superoxide, generated by Hcy, indirectly decreases NO bioavailability by rapid consumption of NO, resulting in the generation of peroxynitrite (ONOO-). Finally, reduction in NO synthesis and its release by injured endothelial cells cause the release of multiple growth factors which provoke proliferation of VSMC.
Hcy can induce apoptosis of endothelial cells by activating the Fas cell-death pathway, the p53/Noxa pathway, and the cytochrome-c activated caspase 3 and 9 pathways which are present in endothelial cells. The initiative step of the atherosclerotic process is the increase in the expression and plasma levels of the inflammatory cytokines, tumour necrosis factor alpha, that enhance the activation of a redox-sensitive nuclear transcription factor, nuclear factor-kappa B in the vasculature [20].
Hcy is also known to decrease NO production by increasing asymmetrical ADMA. This molecule is an endogenous inhibitor of eNOS enzyme [Table/Fig-4]. Defect in DDAHII has been confirmed to be involved in the Hcy-induced dysfunction of endothelial NO system. Jia et al., have reported that hypermethylation of DDAHII contributed to Hcy induced apoptosis of ECs [21]. Also, Zhang et al., have recently reported that Hcy upregulates platelet-derived growth factors levels via DNA demethylation in endothelial cells, that it affects cross-talk between endothelial cells and vascular smooth muscle cells and leads to vascular smooth muscle cells activation [22].
Recently, researchers have paid attention to a connection which was present between homocysteine and an endogenous tryptophan derivative, kynurenic acid. It was revealed that kynurenic acid counteracted the harmful effects of Hcy on endothelium cells in vitro[23]. The contribution of the Hcy-derived methylation modifications to eNOS gene expression seems to be tissue-specific and independent. Hcy-derived methylation modifications to the iNOS gene promoter contribute to a lesser extent to iNOS gene expression [24].
Looking at the big picture, the association between Hcy and endothelial dysfunction depends largely on molecules with damaging effects on eNOS coupling, and loss of NO bioavailability. Hcy was shown to promote the oxidation of the essential eNOS cofactor, BH4, resulting in the uncoupling of the enzyme, timely spontaneous oxygen radical synthesis, and decreased NO production. Besides, this oxidative action is reversed by the supplementation of folic acid and anti-oxidants [25]. In vascular endothelium, 5-methyltetrahydrofolate (5-MTHF) lowers intracellular Hcy and it reduces oxygen radical generation by NADPH-oxidase and iNOS. In addition, 5-MTHF directly scavenges ONOO−, protects the oxidation of BH4, and improves eNOS coupling.
Homocysteine and Endothelial Function
Moderately elevated plasma Hcy levels are highly prevalent in the general population and they are associated with an increased risk for atherosclerosis, which is independent of classic atherosclerotic risk factors [11]. Homocysteine occurs in human blood plasma in several forms, including the most reactive one, homocysteine thiolactone (HTL) – a cyclic thioester, which represents less than <1 % of total plasma Hcy. The increase in extracellular Hcy is toxic to cells and tissues and it has the potential to initiate a broad array of vascular complications [9,10].
Two main pathways of Hcy biotoxicity have been discussed: Hcy-dependent oxidative stress and Hcy-induced protein structure modification, named homocysteinylation. Vascular endothelial cells are very sensitive, even to a mild increase in Hcy concentration. This susceptibility may be explained by the fact that all human endothelial cells do not express the active form of cystathionine β-synthase, and that they are consequently not able to initiate homocysteine catabolism via the transsulfuration pathway. It has been demonstrated that not only Hcy, but that also HTL may modulate the properties and functions of endothelial cells [8].
Oxidative radicals that are required for the development of atherosclerosis are generated by Hcy and they are capable of oxidizing LDL which is present in the plasma (ox-LDL). Recently, researchers have shown that patients with HHcy had an increase in Malondialdehyde (MDA) (end products of lipid peroxidation); this finding was very similar for decreased HDL-associated PON1 enzyme activity, which was associated with increased MDA, to act on endothelial function [26,27]. Herewith, studies have reported that oxidative stress and inhibition of NO release were induced by Hcy, which also promoted a lower expression of PON1 and enhanced the production of ROS and a lower activity of PONs, in patients with HHcy. Hcy-induced ROS downregulates the expression of HDL-associated PON1, which accelerates the development of atherosclerosis [27-29].
PON1 Activity and Endothelial Function
PONs are paralogous enzymes that catalyze the same reaction by using different substrates. The three known mammalian PON families (PON1, PON2, PON3) share a ~ 65% sequence identity, whilst in other vertebrates, a single PON gene is found [30]. The family’s name, paraoxonase, was derived from PON1’s ability to hydrolyze the pesticide, paraoxon [31]. However, in fact, PONs are lactonases. PON1 is a HDL- associated enzyme esterase which appears to contribute to the anti-oxidant and anti-atherosclerotic capabilities of HDL [31].
It has been suggested that improving HDL metabolism, composition, and function was much more efficient than increasing the HDL concentration [11,31]. PON1 has been shown to reduce ROS in human endothelial cells, vascular smooth muscle cells, and fibroblasts [32]. Moreover, recent in vitro studies done, have shown that homocysteinylation of HDL could reduce the activity
of the enzyme, PON1, which was associated with human HDL,
thus rendering the HDL particle more susceptible to oxidative damage [33].
In plasma, Hcy occurs in reduced or oxidized forms, as HTL and as a component of proteins as a result of N- or S-homocysteinylation [33]. The roles of HTL and PON1- lactonase activities as risk factors for the atherosclerosis in cardiovascular disease at the level of endothelial cell, have not been well investigated. Recently, Holven et al., reported that subjects with HHcy had markedly lower PON activities than healthy controls [34].
PON1 exhibits lactonase activity, which prevents LDL oxidation and detoxifies HTL [11]. Since endothelial dysfunction is a very early step in atherogenesis, binding (and subsequent endocytosis) of ox-LDL to endothelial cells is an area of intense investigation. An increased plasma level of ox-LDL is a well-known risk factor for endothelial dysfunction and atherosclerosis. Also, ox-LDL causes activation, followed by dysfunction of endothelium,. Ox-LDL stimulates expression of chemokines and adhesion molecules such as monocyte chemotactic protein-1 (MCP-1), E- and P-selectins, vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) on endothelial cells [35,36]. Experimental studies have shown that ox-LDL causes injury to endothelial cells via activation of different signal transduction pathways such as those involving PKC and MAPK [37-39]. [Table/Fig-5] So, the initial HDL-associated high PON1 activity may reduce the formation of ox-LDL[31]. Besides, PON1 is directly involved in the pathogenesis of atherosclerosis due to modulation of NO bioavailability. Actually, Besler et al., have indicated that HDL-associated PON1 enzyme activity has a major impact on endothelial function, which is consistent with the recorded inverse relationship between PON1 activity and atherosclerotic disease development [39]. It is unknown, whether the loss in PON1 enzyme activity leads to alterations in other HDL constituents, besides MDA, that activates lectin-like ox-LDL receptor-1 (LOX-1) [39-43].
Researchers have shown that LOX-1 was responsible for the binding, internalization and degradation of ox-LDL in endothelial cells [35]. LOX-1 is a scavenger receptor and it is regarded as a central element in the initiation of endothelial dysfunction. Mehta et al., have reported that LOX-1 inhibition was associated with attenuation of atherosclerosis [43]. LOX-1 may be upregulated by its own ligand, ox-LDL or by proinflammatory cytokines in endothelial cells. Also, oxidant species are potent inducers of LOX-1. In vascular endothelial cells, LOX-1 activation has been suggested to induce several intracellular signaling pathways, including protein kinases and transcription factors, which regulate the expressions of genes which are related to atherosclerosis [43,36,44].
Interestingly, factors which modulate the HDL-asssociated PON1 enzyme activity are similar for endothelial function modulation, which encourage the proposal that HDL-asssociated PON1 enzyme activity was the cornerstone for endothelial function. These findings suggest that assays done to study HDL-associated PON 1 enzyme action on endothelium may increase our knowledge to assign atherosclerotic disease risk, and that they may boost our understanding of the outcomes of future trials which test HDL-associated PON1 enzyme targeted therapies. In a recent experimental study, administering small apolipoprotein-mimetic peptides (Apo-A1) to mice was found to reduce atherogenesis [45]. eNOS activation by HDL entails apoA-I–dependent binding of the lipoprotein to scavenger receptor BI (SR-BI) in endothelial cells; this causes a cholesterol efflux that is sensed by SR-BI and it begins a signaling cascade which includes the activation of Src kinases, PI3K, and Akt, which phosphorylates eNOS at Ser1177 to enhance eNOS activity [45,46].
Three types of the enzyme, NOS with different locations and functions
| Nitric oxide synthase | Location and function |
| Type I neural NOS (nNOS) | Brain and peripheral nervous system, retrograde neurotransmitter, memory and learning |
| Type II inducible NOS (iNOS) | Endothelial cells, immune response, defense against pathogens |
| Type III constitutive NOS (cNOS) | Endothelial cells, vazodillatation |
Diseases or conditions associated with abnormal NO production and bioavailability
| Stroke |
| Coronary artery disease |
| Depression |
| Hypertension |
| Obesity |
| Dyslipidemias (particularly hypercholesterolemia and hypertriglyceridemia) |
| Diabetes (both type I and II) |
| Heart failure |
| Atherosclerosis |
| Aging |
| Cigarette smoking |
How unabled is NO through the uncoupled eNOS
Increasing asymmetrical ADMA, and its consequences
List of the ox-LDL-induced intracellular signaling pathways
| p38 (MAPK), |
| p44/42 MAPK , |
| protein kinase C (PKC) , |
| protein kinase B (PKB), |
| protein tyrosine kinase (PTK) |
| transcription factor NF-nB |
| AP-1 |
Conclusion
Elucidating the redox dependent mechanisms which are involved in the physiological and pathophysiological regulations of eNOS by ROS will provide critically new insights into atherosclerotic disease and they may lead to the identification of novel treatment targets which are related to PON 1, Hcy and eNOS-modulated vascular responses. Quite a nice example is the recent study done by Zhang et al., in which they reported that incubation with aged garlic extract preserved normal NO output from endothelial cells, even under conditions of elevated Hcy levels, by increasing cellular thiol antioxidant and prevention of BH4 oxidation.
[1]. P Pacher, JS Beckman, L Liaudet, Nitric oxide and peroxynitrite in health and disease.Physiol Rev. 2007 87(1):315-424. [Google Scholar]
[2]. U Förstermann, T Münzel, Endothelial nitric oxide synthase in vascular disease: from marvel to menace.Circulation. 2006 113(13):1708-14. [Google Scholar]
[3]. S Kawashima, Malfunction of vascular control in lifestyle-related diseases: endothelial nitric oxide (NO) synthase/NO system in atherosclerosis.J Pharmacol Sci. 2004 96(4):411-19. [Google Scholar]
[4]. K Noguchi, N Hamadate, T Matsuzaki, M Sakanashi, J Nakasone, T Uchida, Increasing dihydrobiopterin causes dysfunction of endothelial nitric oxide synthase in rats in vivo. Am J Physiol Heart Circ Physiol. 2011 301(3):721-29. [Google Scholar]
[5]. U Förstermann, WC Sessa, Nitric oxide synthases: regulation and function.Eur Heart J. 2012 33(7):829-37. [Google Scholar]
[6]. NJ Alp, KM Channon, Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease Arterioscler.Thromb. Vasc. Bio. 2004 24:413-20. [Google Scholar]
[7]. CA Gunnett, DD Lund, AK McDowell, FM Faraci, DD Heistad, Mechanisms of Inducible Nitric Oxide Synthase-Mediated Vascular Dysfunction Arterioscler.Thromb. Vasc. Bio. 2005 25:1617-22. [Google Scholar]
[8]. J Malinowska, J Kolodziejczyk, B Olas, The disturbance of hemostasis induced by hyperhomocysteinemia; the role of anti-oxidants.Acta Biochim Pol. 2012 59(2):185-94. [Google Scholar]
[9]. TB Domagała, M Łacinski, WH Trzeciak, B Mackness, MI Mackness, H Jakubowski, The correlation of homocysteine-thiolactonase activity of the paraoxonase (PON1) protein with coronary heart disease status. Cell Mol Biol (Noisy-le-grand). 2006 52(5):4-10. [Google Scholar]
[10]. H Jakubowski, The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease.J Physiol Pharmacol. 2008 59(Suppl 9):155-67. [Google Scholar]
[11]. N Yilmaz, Relationship between paraoxonase and homocysteine: crossroads of oxidative diseases.Arch Med Sci. 2012 8(1):138-53. [Google Scholar]
[12]. MM Steed, SC Tyagi, Mechanisms of cardiovascular remodeling in hyperhomocysteinemia. Antioxid Redox Signal. 2011 15(7):1927-43. [Google Scholar]
[13]. N Tyagi, KS Moshal, AV Ovechkin, W Rodriguez, M Steed, B Henderson, Mitochondrial mechanism of oxidative stress and systemic hypertension in hyperhomocysteinemia.J Cell Biochem. 2005 96(4):665-71. [Google Scholar]
[14]. B Dhillon, MV Badiwala, A Maitland, V Rao, SH Li, S Verma, Tetrahydrobiopterin attenuates homocysteine induced endothelial dysfunction. Mol Cell Biochem. 2003 247(1-2):223-27. [Google Scholar]
[15]. L Rochette, J Lorin, M Zeller, JC Guilland, L Lorgis, Y Cottin, Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol Ther. 2013 140(3):239-57. [Google Scholar]
[16]. L He, H Zeng, F Li, J Feng, S Liu, J Liu, Homocysteine impairs coronary artery endothelial function by inhibiting tetrahydrobiopterin in patients with hyperhomocysteinemia. Am J Physiol Endocrinol Metab. 2010 299(6):E1061-5. [Google Scholar]
[17]. MJ Crabtree, KM Channon, Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease.Nitric Oxide. 2011 25(2):81-8. [Google Scholar]
[18]. A Tawakol, MA Forgione, M Stuehlinger, NM Alpert, JP Cooke, J Loscalzo, Homocysteine impairs coronary microvascular dilator function in humans.J Am Coll Cardiol. 2002 40(6):1051-58. [Google Scholar]
[19]. M Stühlinger, Asymmetric dimethyl arginine (ADMA): a novel cardiovascular risk factor?.Wien Med Wochenschr. 2007 157(3-4):57-60. [Google Scholar]
[20]. P Sharma, RD Senthilkumar, V Brahmachari, E Sundaramoorthy, A Mahajan, A Sharma, Mining literature for a comprehensive pathway analysis: a case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis. 2006 5:1 [Google Scholar]
[21]. SJ Jia, YQ Lai, M Zhao, T Gong, BK Zhang, Homocysteine-induced hypermethylation of DDAH2 promoter contributes to apoptosis of endothelial cells.Pharmazie. 2013 68(4):282-86. [Google Scholar]
[22]. D Zhang, Y Chen, X Xie, J Liu, Q Wang, W Kong, Homocysteine activates vascular smooth muscle cells by DNA demethylation of platelet-derived growth factor in endothelial cells. J Mol Cell Cardiol. 2012 53(4):487-96. [Google Scholar]
[23]. K Wejksza, W Rzeski, WA Turski, Kynurenic acid protects against the homocysteine-induced impairment of endothelial cells.Pharmacol Rep. 2009 61(4):751-56. [Google Scholar]
[24]. M Barroso, MS Rocha, R Esse, JR Gonçalves I, AQ Gomes, T Teerlink, Cellular hypomethylation is associated with impaired nitric oxide production by cultured human endothelial cells. Amino Acids. 2012 42(5):1903-11. [Google Scholar]
[25]. KB Holven, TS Haugstad, T Holm, P Aukrust, L Ose, MS Nenseter, Folic acid treatment reduces elevated plasma levels of asymmetric dimethylarginine in hyperhomocysteinaemic subjects. Br J Nutr. 2003 89(3):359-63. [Google Scholar]
[26]. C Besler, TF Lüscher, U Landmesser, Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease.EMBO Mol Med. 2012 4(4):251-68. [Google Scholar]
[27]. E Eren, N Yilmaz, O Aydin, Functionally Defective High-Density Lipoprotein and Paraoxonase: A Couple for Endothelial Dysfunction in Atherosclerosis. Cholesterol. 2013 2013:792090 [Google Scholar]
[28]. N Yilmaz, O Aydin, A Yegin, A Tiltak, E Eren, G Aykal, Impaired oxidative balance and association of blood glucose, insulin and HOMA-IR index in migraine.Biochem Med (Zagreb). 2011 21(2):145-51. [Google Scholar]
[29]. BA Maron, T Michel, Subcellular localization of oxidants and redox modulation of endothelial nitric oxide synthase.Circ J. 2012 76(11):2497-512. [Google Scholar]
[30]. H Bar-Rogovsky, A Hugenmatter, DS Tawfik, The evolutionary origins of detoxifying enzymes: The mammalian serum paraoxonases (PONs) relate to bacterial homoserine lactonases.J Biol Chem. 2013 288(33):23914-27. [Google Scholar]
[31]. E Eren, N Yilmaz, O Aydin, High Density Lipoprotein and it’s Dysfunction.Open Biochem J. 2012 6:78-93. [Google Scholar]
[32]. S Horke, I Witte, P Wilgenbus, M Krüger, D Strand, U Förstermann, Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation.Circulation. 2007 115(15):2055-64. [Google Scholar]
[33]. J Perła-Kaján, H Jakubowski, Paraoxonase 1 and homocysteine metabolism. Amino Acids. 2012 43(4):1405-17. [Google Scholar]
[34]. KB Holven, H Scholz, B Halvorsen, P Aukrust, L Ose, MS Nenseter, Hyperhomocysteinemic subjects have enhanced expression of lectin-like oxidized LDL receptor-1 in mononuclear cells. J Nutr. 2003 133(11):3588-91. [Google Scholar]
[35]. S Mitra, T Goyal, JL Mehta, Oxidized LDL, LOX-1 and atherosclerosis.Cardiovasc Drugs Ther. 2011 25(5):419-29. [Google Scholar]
[36]. S Mitra, A Deshmukh, R Sachdeva, J Lu, JL Mehta, Oxidized low-density lipoprotein and atherosclerosis implications in antioxidant therapy. Am J Med Sci. 2011 342(2):135-42. [Google Scholar]
[37]. SA Abdelsamie, Y Li, Y Huang, MH Lee, RL Klein, G Virella, Oxidized LDL immune complexes stimulate collagen IV production in mesangial cells via Fc gamma receptors I and III. Clin Immunol. 2011 139(3):258-66. [Google Scholar]
[38]. HH Wang, HL Hsieh, CY Wu, CM Yang, Oxidized low-density lipoprotein-induced matrix metalloproteinase-9 expression via PKC-delta/p42/p44 MAPK/Elk-1 cascade in brain astrocytes. Neurotox Res. 2010 17(1):50-65. [Google Scholar]
[39]. C Besler, K Heinrich, L Rohrer, C Doerries, M Riwanto, DM Shih, Mechanisms underlying adverse effects of HDL on eNOS-activating pathways in patients with coronary artery disease.J Clin Invest. 2011 121(7):2693-708. [Google Scholar]
[40]. C Besler, TF Lüscher, U Landmesser, Molecular mechanisms of vascular effects of High-density lipoprotein: alterations in cardiovascular disease.EMBO Mol Med. 2012 4(4):251-68. [Google Scholar]
[41]. J Barlic, PM Murphy, An oxidized lipid-peroxisome proliferator-activated receptor gamma-chemokine pathway in the regulation of macrophage-vascular smooth muscle cell adhesion.Trends Cardiovasc Med. 2007 17(8):269-74. [Google Scholar]
[42]. L Calabresi, M Gomaraschi, G Franceschini, Endothelial protection by highdensity lipoproteins: from bench to bedside.Arterioscler Thromb Vasc Biol. 2003 23(10):1724-31. [Google Scholar]
[43]. JL J Mehta, J Chen, PL Hermonat, F Romeo, G Novelli, Lectin-like, oxidized lowdensity lipoprotein receptor-1 (LOX-1): A critical player in the development of atherosclerosis and related disorders.Cardiovasc Res. 2006 69(1):36-45. [Google Scholar]
[44]. R Yoshimoto, Y Fujita, A Kakino, S Iwamoto, T Takaya, T Sawamura, The discovery of LOX-1, its ligands and clinical significance.Cardiovasc Drugs Ther. 2011 25(5):379-91. [Google Scholar]
[45]. MJ Amar, W D’Souza, S Turner, S Demosky, S Sviridov, D Stonik, 5A apolipoprotein mimetic peptide promotes cholesterol efflux and reduces atherosclerosis in mice. J Pharmacol Exp Ther. 2010 334(2):634-41. [Google Scholar]
[46]. C Mineo, PW Shaul, Role of high-density lipoprotein and scavenger receptor B type I in the promotion of endothelial repair. Trends Cardiovasc Med. 2007 17(5):156-61. [Google Scholar]