Unusual Case of Vogt-Koyanagi-Harada Syndrome Presenting as Non-specific Headache
Pradeep A.V.1, Arun Kumar J.S.2, Naveen K.N.3, Sonali Rao4, Sharan Shetty5
1 Assistant Professor, Department of Ophthalmology, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & HospitalSattur, Dharwad Karnataka, India.
2 Professor, Department of ENT, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & HospitalSattur, Dharwad Karnataka, India.
3 Associate Professor, Department of Dermatology, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & HospitalSattur, Dharwad Karnataka, India.
4 Post Graduate, Department of Ophthalmology, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & HospitalSattur, Dharwad Karnataka, India.
5 Post Graduate, Department of Ophthalmology, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & HospitalSattur, Dharwad Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Pradeep A.V., Assistant Professor, Department of Ophthalmology, Sri Dharmasthala Manjunatheshwara College of Medical Sciences & Hospital Sattur, Dharwad Karnataka-580009, India.
Phone: 09916915716,
E-mail: dr.avpradeep@gmail.com
Vogt–Koyanagi–Harada syndrome (VKH) is a bilateral intraocular granulomatous panuveitis which is frequently associated with systemic manifestations such as meningismus, tinnitus, poliosis and vitiligo of autoimmune aetiology. Headache by itself, does not fulfill the diagnostic criteria and is insufficient for the diagnosis. A 22-year-old male presented with a 10 day history of headache, followed by decreased vision in both eyes. Slit lamp biomicroscopy revealed sluggishly reactive pupils with anterior uveitis, mild vitritis and hyperaemic discs with bilateral exudative retinal detachments. All uveitis workups were negative. Follow up of three years revealed no neurological or auditory symptoms. Headache alone, followed by decreased vision, before the onset of neurological and auditory symptoms, can be an initial presentation of Vogt-Koyanagi-Harada (VKH) syndrome. VKH should be considered in the differential diagnosis of atypical presentations of headache.
Headache, Panuveitis, Vogt–Koyanagi–Harada syndrome
Case Report
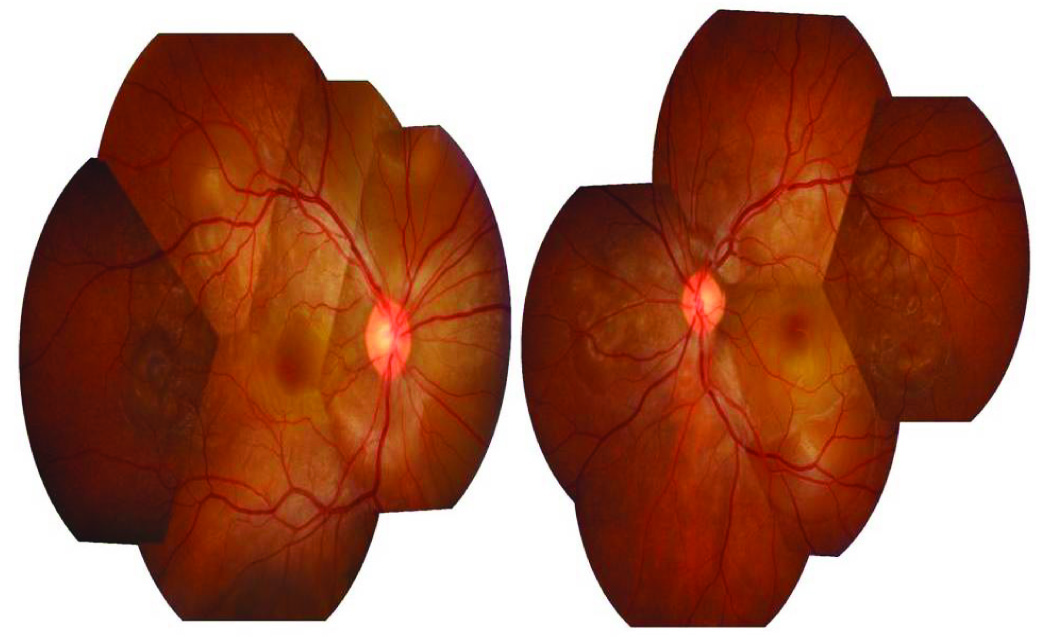
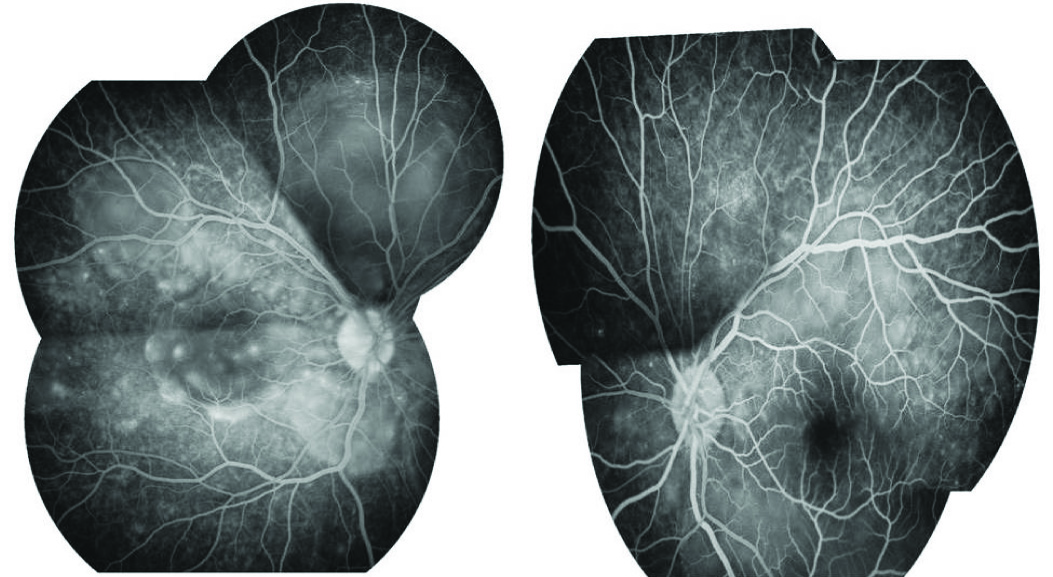
A 22-year-old south Indian male who did not have any medical illnesses in the past, presented with a 10 day history of headache. He was evaluated at a general clinic and was treated symptomatically. Seven days later, he presented with decreased vision in both eyes, along with vomiting, to the Ophthalmology Clinic. Headache was severe and throbbing in nature. It was not associated with fever, ataxia, neck stiffness or cerebellar signs. There was no history of any previous surgery or ocular trauma. There was no history of tinnitus, alopecia, vitiligo, poliosis, weakness of limbs, backpain, numbness or joint pain. At presentation, visual acuities were recorded as 1/60 and 1/120 in the right eye and left eye, respectively. Anterior segment examination showed bilateral sluggishly reactive pupils of sizes 4 mm and 3 mm in right eye and left eye respectively. Anterior chamber showed 2+ cells in both eyes, with no keratic precipitates. Intraocular pressure in each eye was within normal limits. Vitreous showed 1+ cells, which indicated mild vitritis. Fundus examination showed hyperaemic discs in both eyes, with multiple exudative retinal detachments [Table/Fig-1]. A fundus fluorescein angiogram (FFA) showed multiple areas of pinpoint hyperfluorescent spots at the level of retinal pigment epithelium, with subsequent leakage and pooling of dye in the subretinal spaces of both eyes, along with leakage and staining of the discs in the late phase [Table/Fig-2]. Computed tomography (CT) scan of the brain and chest X-ray were unremarkable. Complete haemogram, serum electrolyte levels, which were assessed, were found to be within normal limits. Investigations which included tuberculin skin tests, Venereal Diseases Research Laboratory (VDRL) test, Dengue serology, malaria card test were negative. Cerebrospinal fluid (CSF) analysis predominantly showed lymphocytes (90%) and neutrophils (10%) with CSF sugar of 51 mg% (50-80mg %) and CSF protein of 89 mg% (10-45mg %), which indicated pleocytosis. An audiological evaluation done was within normal limits. A skin biopsy was taken and it was reported as normal. A comprehensive assessment was done to rule out other causes of uveitis.
Montage fundus photo showing bilateral hyperemic discs with multiple serous retinal detachments
FFA showing bilateral multiple pinpoint leakages with pooling of dye and staining of the discs in the late phase
The patient was diagnosed as an incomplete case of VKH syndrome and he was started on oral prednisolone (75 mg/day) for 1 week, with a weekly taper of 5 mg, along with 1% topical prednisolone acetate, six times a day and 1% topical cyclopentolate three times a day. There was a marked improvement in headache after 1 week of treatment and the vision improved to 20/20 in each eye after 4 weeks.
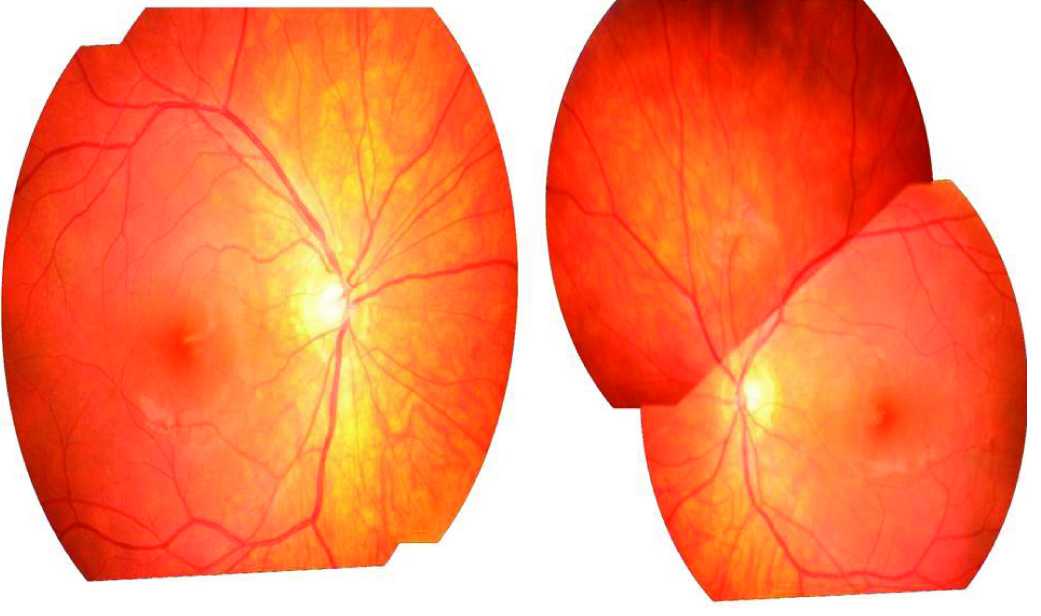
Six months later, the patient presented again with headache but without visual complaints. On examination, anterior segment was found to be clear, with mild vitritis. There was no history of any extraocular manifestation. He was restarted on oral steroids with topical prednisolone acetate. There was a marked improvement in his headache within one week, with sunset glow of fundus, bilaterally [Table/Fig-3].
Montage photo showing bilateral ‘sunset glow’ fundus six months later
The patient was found to be asymptomatic at the end of a three year follow up, without having skin or audiological manifestations.
Discussion
The VKH syndrome is clinically divided into: a prodromal phase which consists of meningeal symptoms, an acute uveitic phase which is characterized by ocular and auditory involvement, a convalescent phase which comprises of characteristic retinal changes along with skin manifestations and a chronic recurrent phase [1,2] of all the patients who present with uveitis in India, 1 in 50 have VKH syndrome [3]. A majority of the patients present with neurological signs, blurring of vision and extraocular manifestations, with characteristic ocular signs. The ocular presentation begins with bilateral diffuse granulomatous panuveitis, hyperaemic discs, exudative retinal detachments and gradual depigmentation of the choroid, which lead to typical sunset glow of fundus [4]. According to the revised diagnostic criteria set for VKH syndrome [2] one of the criteria for its diagnosis is meningismus, which is characterized by fever, headache, malaise, nausea, stiffness of neck and back. Headache alone is not adequate for diagnosing VKH. Though headache is a common symptom, it is usually associated with blurring of vision in most of the cases at presentation. Literature reports show that only few cases of VKH syndrome present with headache as an early symptom. Cho et al., had described a thunderclap headache which mimicked pituitary apoplexy [5]. Khairallah et al., reported one case of VKH with diffuse headache, which was followed by decreased vision. The patient subsequently developed complete VKH syndrome [6]. Kosama et al., also reported a patient with diffuse headache, who developed neurological and audiological symptoms [7]. Tavsanli et al described 2 cases of VKH syndrome which presented with severe headache, along with decreased vision, which gradually improved with course of steroids [8]. Our case presented with non-specific headache initially, which was treated symptomatically, until the patient presented with sudden blurring of vision. The patient subsequently developed retinal detachments and a sunset glow of fundus within a few months and he did not develop any audiological or skin manifestations during 3 year’s follow-up. The patient was started on oral steroids and there was an improvement in his vision within a week. He was labeled as incomplete VKH syndrome. The recurrent attack was again promptly controlled by oral steroids. The above case illustrates that non-specific headache can be one of the early manifestations of VKH syndrome.
Conclusion
It is imperative for physicians and neurologists to recognize this as one of the differential diagnoses of headache.
A timely referral to an ophthalmologist, along with a prompt intervention, can give patients excellent visual outcomes and prevent long term sequelae.
[1]. Moorthy RS, Inomata H, Rao NA, Vogt–Koyanagi–Harada syndromeSurv Ophthalmol 1995 39(4):265-92. [Google Scholar]
[2]. Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L, Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclatureAm J Ophthalmol 2001 131(5):647-52. [Google Scholar]
[3]. Mondkar SV, Biswas J, Ganesh SK, Analysis of 87 cases with Vogt-Koyanagi-Harada diseaseJpn J Ophthalmol 2000 44(3):296-301. [Google Scholar]
[4]. Naeini AE, Daneshmand D, Khorvash F, Chitsaz A, Vogt-Koyanagi-Harada syndrome presenting with encephalopathyAnn Indian Acad Neurol 2013 16(2):264-5. [Google Scholar]
[5]. Cho JH, Ahn JY, Byeon SH, Huh JS, Thunderclap headache as initial manifestation of Vogt–Koyanagi–Harada diseaseHeadache 2008 48(1):153-5. [Google Scholar]
[6]. Khairallah A.S., Headache as an initial manifestation of Vogt–Koyanagi–Harada diseaseSaudi J Ophthalmol 2013 http://dx.doi.org/10.1016/j.sjopt.2013.10.003 [Google Scholar]
[7]. Kosma KK, Kararizou E, Markou I, Eforakopoulou E, Kararizos G, Mitsonis C, Headache as a first manifestation of Vogt–Koyanagi–Harada diseaseMed Princ Pract 2008 17(3):253-4. [Google Scholar]
[8]. Tavsanli M, Uluduz D, Saip S, Kendiroglu G, Vogt–Koyanagi–Harada disease: headache as an initial manifestationJ Headache Pain 2008 9(4):255-6. [Google Scholar]