Haemophagocytic lymphohistiocytosis (HLH) is a potentially fatal, hyper inflammatory condition which is caused by a highly stimulated but ineffective immune response. We are presenting here, a case of HLH which occurred in a 45 day infant. Presence of lung cavity and a lytic bone lesion in the skull, as was seen in this case, have not been reported in HLH in the literature. This raises a possibility of a simultaneous occurrence of HLH and Langerhans cell histiocytosis. In a child who presents with septicaemia but does not respond to treatment, the possibility of HLH needs to be considered.
Haemophagocytic lymphohistiocytosis, Familial haemophagocytic lymphohistiocytosis, Lung cavity
Case Report
A 45-day-old baby boy presented with a history of fever, cough and cold since 15 days, breathing difficulty and refusal to feed since one day. The child had been hospitalized for five days for the same and had received multiple antibiotics. Radiograph and CT of the chest done during that admission showed a cavity in the right middle lobe. The baby was born of a third degree consanguineous marriage. There was a history of death of an older female sibling at age of two months and a spontaneous abortion at five months of gestation. The eldest male child was alive and well. The child was exclusively breast fed and was immunized as per schedule. On admission, he had respiratory distress with tachypnoea and tachycardia. Oxygen saturation on room air was 76%. The child was febrile, pale and had liver enlarged four cm below the right costal margin (span eight cm) and spleen enlarged two cm below the left costal margin. He had bilateral crepitations and rhonchi, on respiratory examination.
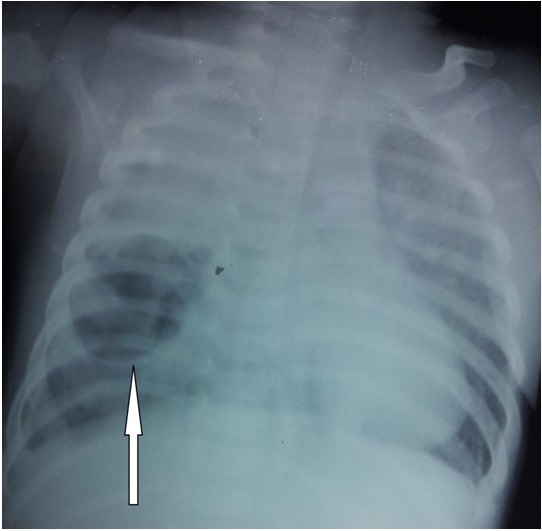
Initial investigations showed haemoglobin of 8 g%, total leukocyte count (TLC) of 1000/cmm with 65 % neutrophils and a platelet count of 60000/cmm. Aspartate transaminase was 478 (Normal 15 to 55 IU/l) and Alanine transaminase was 86 (Normal 5-45 IU/l). Serum bilirubin and renal function tests were normal. Prothrombin time showed an INR of 1.4. The chest radiograph showed a cavity of size about 3 cm by 3 cm in the right middle lobe and a homogenous opacity in the right upper lobe [Table/Fig-1] .
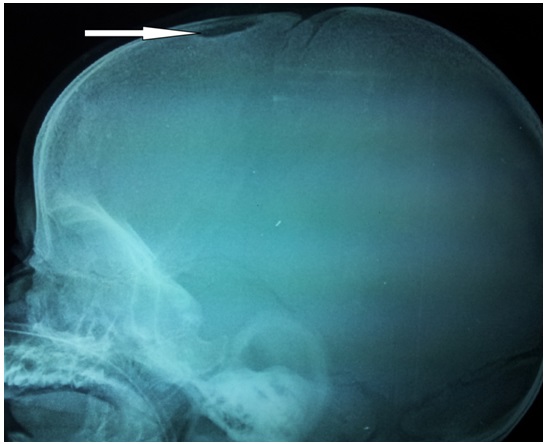
The child was ventilated and antibiotics were started. An ultrasound of the abdomen showed hepatosplenomegaly. C reactive protein, blood culture and bronchoscopy were normal. The bronchoalveolar lavage was negative for gram/acid fast bacilli/fungal stains as well as cultures. Repeat investigations revealed a TLC of 900/cmm and a platelet count of 35000/cmm. Serum triglycerides were elevated to 380 mg % (Normal upto 170mg %). Serum ferritin levels and serum fibrinogen levels were 1752.22 ng/ml (Normal range 25 to 400 ng/ml) and 106.1 mg/dl (Normal range 180-350mg/dl) respectively. LDH was raised to 1568 (Normal Range 170-580 U/l). Hence, a diagnosis of HLH was made on the basis of fulfilment of 5 out of 8 criteria for haemophagocytic lymphohistiocytosis and the child was started on dexamethasone. The child also developed a nodular lesion of 0.5 cm x 0.5 cm on the scalp after admission. Radiograph of the skull revealed a punched out lesion of about 1cm x 1cm [Table/Fig-2] . Bone marrow / liver biopsy and biopsy of the skull lesion could not be done due to poor general condition of the patient. The child died within 12 hours of starting the treatment.
Chest radiograph showing lung cavity
Radiograph of skull showing lytic lesion
Discussion
Haemophagocytic Lymphohistiocytosis (HLH) is a potentially fatal hyper inflammatory condition which is caused by a highly stimulated but ineffective immune response [1] . HLH is associated with a variety of underlying conditions and it is characterized by proliferation and activation of macrophages in the reticuloendothelial system [2] .
HLH can be classified according to the underlying aetiology into either primary (genetic) or secondary (acquired) HLH [1] . Both can occur at any age and they can be precipitated by infections [2] . Primary HLH can further be divided into two subgroups: Familial Haemophagocytic Lymphohistiocytosis (FHL) and specific immune deficiencies like Chédiak-Higashi syndrome 1, etc, [3] . The onset of disease in FHL occurs below one year of age in a majority of the cases. Acquired (secondary) forms of HLH can occur in all age groups [3] . It occurs most commonly in the setting of infections, with viruses of the herpes group being the commonest triggers [2] .Although it was first described in 1952, a set of diagnostic guidelines for HLH was first presented by the Histiocyte Society in 1991 and it was revised in 2004 [4] .
HLH-2004 diagnostic criteria
The diagnosis of HLH can be established if either 1 or 2 of the below criteria is/are fulfilled:
A molecular diagnosis which is consistent with HLH is made.
Diagnostic criteria for HLH are fulfilled (5 of the 8 criteria below):* Fever Splenomegaly Cytopaenias (affecting 2-3 lineages in the peripheral blood):
haemoglobin < 90 g/L (in infants who are < 4 weeks of age, haemoglobin < 100 g/L), platelets < 100 x 109/L, neutrophils <1.0 x 109/L Hypertriglyceridaemia and/or hypofibrinogenaemia: fasting triglycerides > 3.0 mmol/L (i.e., > 265 mg/dL), fibrinogen < 1.5 g/L Haemophagocytosis in BM, spleen, or lymph nodes A low or absent NK-cell activity (according to local laboratory references) Ferritin < 500 ng/L Soluble CD25 (i.e., sIL2r)<2400 U/mL *Supportive criteria include neurologic symptoms, cerebrospinal fluid pleocytosis, conjugated hyperbilirubinaemia and transaminitis, hypoalbuminaemia, hyponatraemia, elevated D-dimers, and actate dehydrogenase. The absence of haemophagocytosis in the BM does not exclude a diagnosis of HLH. 5 of 8 criteria are required to make the diagnosis of HLH. Haemophagocytosis, a hallmark of activated macrophages, is neither specific nor sensitive for HLH and its presence should only be considered as supportive [1] .
Clinical manifestations of HLH include prolonged fever, hepatosplenomegaly, bleeding, skin rash, CNS abnormalities, jaundice, and laboratory findings of bicytopaenia or pancytopaenia, coagu lopathy,hyperlipidaemia,hypofibrinogenaemia,hyperferritinaemi a, etc. The cardinal symptoms of HLH are prolonged high fever, hepatosplenomegaly and cytopaenias [5] . Ramchandran and coworkers, in their case series of HLH, reported fever with or without additional symptoms or signs such as rash, hepatosplenomegaly, lymphadenopathy, respiratory symptoms [cough, tachypnoea, dyspnoea] or CNS involvement (irritability, altered behavior, meningeal signs or seizures), as reasons for hospital admissions. Fever with hepatomegaly was the most common presentation at admission, while respiratory symptoms were seen in nearly 30% of the patients in this series [6] . Similarly, the case series from B J Wadia Hospital for children, Mumbai also had fever and hepatosplenomegaly as the most common presentations [7].
The signs and symptoms of HLH are often non specific and hence making a diagnosis is difficult [8] . Many patients with severe sepsis, systemic inflammatory response syndrome and multiple organ dysfunction syndrome may also meet the diagnostic criteria for HLH [4] . HLH should be considered in the differential diagnosis of children with sepsis or presumed sepsis that do not respond to the conventional treatment [6] .
The treatment of HLH should include the suppression of the severe hyper inflammation, the killing of pathogen-infected cells, treatment to restore normal organ function, and treatment of underlying diseases, i.e., stem cell transplantation for patients with genetic disorders [4] .
The present case had fever, hepatosplenomegaly and respiratory distress at admission. However, it did not respond to treatment and it fulfilled the criteria for diagnosis as HLH. As attempts made to distinguish primary and secondary haemophagocytic lymphohistiocytosis in children have been inconclusive, a unifying diagnosis of HLH is appropriate [9] .
The differential diagnosis of a cavitatory lung lesion at this age includes cavitating granuloma, congenital bronchopulmonary malformation, hyper IgE syndrome, Langerhans cell histiocytosis, postinfectious (Staphylococcal), septic emboli, etc [10] . Lung cavity or lytic bone lesion as a feature of HLH was not found to be described anywhere in the literature. These two findings raise the possibility of an overlap between HLH and Langerhans cell histiocytosis (LCH). Few case reports described both entities in the same patient, but none mentioned them at such an early age [11-13] . The association between LCH and haemophagocytic syndrome has not been well established, but T-lymphocytes and cytokines are prominent in the pathogenesis of both disorders and they are presumed to link the two [13] .
To conclude, the possibility of HLH should be considered in a child who presents with septicaemia which does not respond to treatment. Easily available tests like serum ferritin and serum triglycerides may provide useful clues for diagnosing this condition.
[1]. S Weitzman, Approach to hemophagocytic syndromesHematology Am Soc Hematol Educ Program. 2011 2011:178-83. [Google Scholar]
[2]. M Szyper-Kravitz, The hemophagocytic syndrome/macrophage activation syndrome: a final common pathway of a cytokine storm.Isr Med Assoc J. 2009 11:633-4. [Google Scholar]
[3]. GE Janka, Familial and acquired hemophagocytic lymphohistiocytosis.Eur J Pediatr 2007 166:95-109. [Google Scholar]
[4]. YM Tang, XJ Xu, Advances in hemophagocytic lymphohistiocytosis: pathogenesis, early diagnosis/differential diagnosis, and treatment.Scientific World Journal. 2011 11:697-708. [Google Scholar]
[5]. G Janka, U Zur Stadt, Dental and oral discolorations associated with minocycline and other tetracycline Familial and acquired hemophagocytic lymphohistiocytosis.Hematology Am Soc Hematol Educ Program 2005 :82-8. [Google Scholar]
[6]. B Ramachandran, S Balasubramanian, N Abhishek, KG Ravikumar, AV Ramanan, Profile of hemophagocytic lymphohistiocytosis in children in a tertiary care hospital in India.Indian Pediatr 2011 48:31-5. [Google Scholar]
[7]. R Joshi, A Phatarpekar, Z Currimbhoy, M Desai, Hemophagocytic lymphohistiocytosis: a case`series from MumbaiAnn Trop Paediatr 2011 31:135-40.doi:10.1179/1465328111Y.0000000009. [Google Scholar]
[8]. M Ferreira, J Martins, C Silvestre, C Abadesso, E Matias, H Loureiro, Familial haemophagocytic lymphohistiocytosis: two case reports.BMJ Case Rep 2010 doi:10.1136/bcr.11.2009.2463. [Google Scholar]
[9]. DS Morrell, MA Pepping, JP Scott, NB Esterly, BA Drolet, Cutaneous manifestations of hemophagocytic lymphohistiocytosis.Arch Dermatol. 2002 138:1208-12. [Google Scholar]
[10]. B Newman, EL Effmann, Lung masses. In: Slovis TL, editor. Caffey’s Pediatric Diagnostic Imaging, 2008 11th EditionPhiladelphiaMosby/Elsevier Publishers:1294-323.. [Google Scholar]
[11]. A Klein, F Corazza, A Demulder, D van Beers, A Ferster, Recurrent viral associated hemophagocytic syndrome in a child with Langerhans cell histiocytosisJ Pediatr Hematol Oncol 2008 21:554-56. [Google Scholar]
[12]. PB Hesseling, G Wessels, RM Egeler, DJ Rossouw, Simultaneous occurrence of viral-associated hemophagocytic syndrome and Langerhans cell histiocytosis: a case report.Pediatr Hematol Oncol 1995 12:135-41. [Google Scholar]
[13]. ET Saribeyoglu, S Anak, L Agaoglu, O Boral, A Unuvar, O Devecioglu, Secondary hemophagocytic lymphohistiocytosis induced by malaria infection in a child with Langerhans cell histiocytosis.Pediatr Hematol Oncol 2004 21:267-72. [Google Scholar]