Adrenocortical Carcinoma Presenting as A Rupture and Extensive Retroperitoneal Haemorrhage
Sunil Vitthalrao Jagtap1, Sushama Desai2, Sandeepan Halder3, Swati S. Jagtap4, Anuya Shrikant Badwe5
1 Associate Professor, Department of Pathology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
2 Professor, Department of Pathology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
3 Assistant Lecturer, Department of Physiology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
4 Associate Professor, Department of Physiology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
5 Assistant Lecturer, Department of Pathology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Sunil Vitthalrao Jagtap, Associate Professor, Department of Pathology, Krishna Institute of Medical Sciences, KIMS University, Karad, India.
Phone: 9960628672,
E-mail: drsvjagtap@gmail.com
Adrenocortical carcinoma (ACC) is an extremely rare tumour. We are reporting a 45-year-male patient who had a history of sudden severe worsening epigastric pain and fullness in abdomen, with giddiness. The radiological investigations showed a large right suprarenal mass with extensive destruction and retroperitoneal haemorrhage, with extra capsular, periportal and liver metastases. Exploratory laparotomy was done for excisions of mass and surrounding tissue. On histopathological examination, diagnosis was given as Adreno Cortical Carcinoma with capsular, vascular, and soft tissue nodular involvement.
Adreno-cortical carcinoma, Suprarenal mass, ACC with metastasis
Case 1
A 45-year-old male patient presented with a history of severe pain in abdomen, in the epigastric region, with fullness in abdomen and associated giddiness. On examination, pallor, sweating, bradycardia, and hypotension were noted. Radiological examinations (Ultra Sonography and CT scan of abdomen and pelvis) showed a large right suprarenal mass which measured approximately 15x8.5x8cms, which extended upto right crus of diaphragm superiorly, invaded inferior vena cava and extended over right renal upper pole, displacing it posterior and inferiorly. Retroperitoneal haemorrhage and multiple nodular lesions in right hepatic lobe were also noted. A radiological diagnosis of a suprarenal mass, ACC, Renal Cell Carcinoma, Pheochromocytoma was given.
Patient was asymptomatic before this manifestation. He had no history of diabetes mellitus, hypertension, tuberculosis or any other major systemic illnesses. His haematological and biochemical investigations were within normal limits. His urine Vinyl Mandellic Acid was 5.9 mg/day (normal value 2-14mg/day). His hormonal evaluation was normal. Fasting blood sugar level was 90 mg/dl, serum potassium level was 4.1mEq/L. Liver and renal function tests were within normal limits. Electrocardiography showed normal picture. Emergency exploration and excision of right suprarenal mass was done, along with excisions of adjacent thickened fibroadipose tissue and nodular lesion at paracaval and periportal regions. The underlying kidney was displaced posterio-inferiorly, with no evidence of tumour invasion.
Gross Features
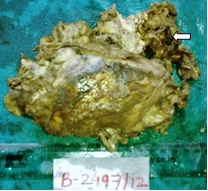
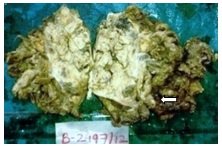
The gross specimen consisted of large, pink, yellow-brown, friable mass which measured 13x9x8 cms and weighed 350gms, with destruction of capsule [Table/Fig-1]. On cut section, it showed a variegated tumour with intratumoural tan yellow nodules [Table/Fig-2]. Larger nodule measured 2.2x2x1.8cms. Tumour showed areas of haemorrhage and necrosis. Adherent fibro-fatty tissue showed nodular lesions.
Gross specimen of suprarenal mass with Irregular capsular destruction and haemorrhage (arrow)
Cut section of mass showed yellowish brown to variegated tumor with areas of haemorrhage and necrosis (arrow)
Light Microscopy
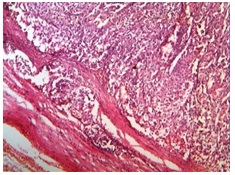
Multiple sections from the mass were taken up for histopathological studies.Histopathology showed a tumour composed of cells which were arranged in large sheets, which was interrupted by fine sinusoidal network, nests, and in areas, an alveolar pattern, with areas of capsular invasion [Table/Fig-3].
Photomicrograph of Adrenocortical carcinoma showing capsular invasion (H&E stain, x100)
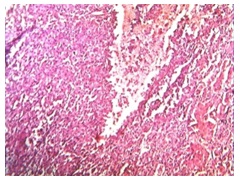
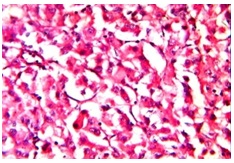
Tumour showed diffuse pattern with extensive areas of haemorrhage [Table/Fig-4] and necrosis, with yellowish and brown pigments (haemosiderin). Individual cells were round to polygonal, which had mild to moderately pleomorphic, hyperchromatic or vesicular nuclei with occasional prominent nucleoli and moderate amount of cytoplasm. In areas, clear cells were seen, which were less than 15%. Few cells showed moderate to severe pleomorphism and in some areas, mitotic activity (5- 6 per 10 HPF) was noted [Table/Fig-5]. Adherent surrounding fibrofatty tissue showed foci of tumour metastasis in our case.
Tumor with areas of haemorrhage (H&E stain x100)
Tumor arranged in diffuse pattern, nests. Individual cells with pleomorphism and increased mitoses (H&E stain x400)
Discussion
ACC is an extremely rare malignancy which has an estimated annual incidence of between 1.5-2 per million population [1]. It is an aggressive malignant neoplasia with poor prognosis. Non functioning tumours are predominant in men in the 4th to 5th decades of life [2,3]. Aetiology is unknown. A majority of cases are incidentally diagnosed at advanced stages with metastasis, because of the tendency of the tumour to invade vascular structures. Most patients of ACC are adults with an average age of about 50 years; many paediatric cases have also been noted [4]. This tumour can be found incidentally at autopsy or as a result of radiological investigations. It may be detected because of signs and symptoms which are related to hormonal dysfunction (about 50%) or which may be evident because of mass effect [5]. ACC is remarkable for many hormonal syndromes which can occur in patients with steroid hormone producing (functional) tumours, that include Cushing’s syndrome, Conn’s syndrome, virilization and feminization. In our case, tumour was non functional. Non functional variants of ACC are hormonally silent tumours which account for approximately 40% of patients. Some cases present with abdominal pain or fullness or symptoms which are related to metastasis [6]. These tend to be more common in older patients and they progress rapidly than functional tumours. Unfortunately, most patients present with metastases at the time of diagnosis, most frequently to the regional lymph nodes in the retroperitoneum, lungs and bones [3]. In our case, patient was asymptomatic till this event. On radiological investigation, our patient was found to have a large right suprarenal mass with rupture, haemorrhage, a local nodular lesion at the hilum, paracaval lymphadenopathy and metastatic nodular lesion in the liver. There was no involvement of pancreas, kidney and brain on CT scan. Presentation of tumour as rupture of suprarenal mass with retroperitoneal haemorrhage is rare. Similar findings were also reported by Sakata R et al., [7] Suyama K et al., [8].
The diagnosis of ACC on histologic examination requires a thorough sampling of the specimen. Several authors have proposed various criteria, both histologic and non histologic, for adrenal cortical neoplasms. Weiss [9] and his colleagues proposed the presence of three or more of the following nine histologic criteria as effective predictors of malignancies: nuclear grade, a mitotic rate of more than 5/50 high power field, abnormal mitoses, less than 25% clear cells, more than 1/3rd diffuse architecture , necrosis, venous invasion and capsular invasion. Out of nine, three criteria were required to label the tumour as malignant. Our case fulfilled almost all these criteria.
It is often necessary to differentiate adrenocortical carcinomas from other conditions like renal cell carcinoma, adrenocortical adenoma and phaeochromocytoma. In our case, renal cell carcinoma was ruled out on radiological and intraoperative findings, as there was no evidence of renal involvement by the tumour. Adrenocortical adenoma was ruled out on histopathology by applying Weiss’ histologic criteria.
ACCs show positivities on immunostaining for Vimentin, Synaptophysin, Melanin-A, Calretinin, Inhibin and this is important for separating adrenal cortical neoplasms from pheochromocytomas [10]. The prognosis is poor, with a significant proportion of patients having distant metastases at the time of their presentations.
Conclusion
In conclusion, we are reporting an extremely rare case of nonfunctional ACC, which presented with rupture and extensive retroperitoneal haemorrhage. This lesion is often difficult to distinguish from other suprarenal masses. Radiographic imaging, a thorough and careful histological study, have significant roles in diagnosis and management of ACC.
[1]. Lombardi G, Rossi R, Adreno cortical carcinoma :12 years prospective experienceWord J Surg 2004 28:896-903. [Google Scholar]
[2]. Icard P, Chapuis Y, Adrenssian B, Bernard A, Proye C, Adreno cortical carcinoma in surgically treated patients. A retrospective study on 156 cases by the French Association of Endocrine surgerySurg 1992 112:972-79. [Google Scholar]
[3]. Ng L, Libertino JM, Adrenocortical carcinoma: diagnosis, evaluation and treatmentJ Urol 2003 Jan 169(1):5-11. [Google Scholar]
[4]. Kay R, Schumacher OP, Tank ES, Adreno cortical carcinoma in childrenJ Urol 1983 130:1130-32. [Google Scholar]
[5]. Mantero F, Terzolo M, Arnaldi G, Osella G, A survey on adrenal incidentaloma in ItalyJ Clin Endocreniol Metab 2000 85:637-44. [Google Scholar]
[6]. Wajchenberg BL, Albergaria Pereira MA, Medonca BB, Adrenocortical carcinoma: Clinical and laboratory observationsCancer 2000 88(4):711-36. [Google Scholar]
[7]. Sakata R, Tsuchiya F, Osaka K, Fujikawa A, Adrenocortical carcinoma detected by retroperitoneal haemorrhageHinyokika Kiyo 2012 Mar 58(3):149-53. [Google Scholar]
[8]. Suyama K, Beppu T, Isiko T, Sugiyama S, Matsumoto K, Spontaneous rupture of adrenocortical carcinomaAm J Surg 2007 194(1):77-78. [Google Scholar]
[9]. Weiss LM, Medeiros LJ, Vickery AL. Jr, Pathologic Features of prognostic significance in adreno cortical carcinomaAm J Surg Pathol 1989 13:202-06. [Google Scholar]
[10]. Jorda M, De MB, Calretinin and Inhibin A are useful in separating adrenocortical neoplasm from pheochromocytomaAppl Immunohistochemistry Mol Morpho 2002 10:67-70. [Google Scholar]