Juvenile Hyaline Fibromatosis (JHF): A Rare Case with Recurrence
Rashmi M.V1, Geetha J.P2, Srinivas Arava3, Niranjana Murthy B4, Kodandaswamy C.R5
1Associate Professors, Department of Pathology,Sri Siddhartha Medical College, Tumkur, Karnataka, India.
2Associate Professors, Department of Pathology,Sri Siddhartha Medical College, Tumkur, Karnataka, India.
3Professor, Department of Surgery,Sri Siddhartha Medical College, Tumkur, Karnataka, India.
4Professors, Department of Pathology,Sri Siddhartha Medical College, Tumkur, Karnataka, India.
5Professors, Department of Pathology,Sri Siddhartha Medical College, Tumkur, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rashmi M.V., Associate Professor, Department of Pathology, Sri Siddhartha Medical College, Agalakote, Tumkur-572104, Karnataka, India.
Phone: 09449666400,
E-mail: rashmiarava@yahoo.com
Juvenile hyaline fibromatosis (JHF) is a rare autosomal recessive disease. Less than 70 cases of JHF have been reported worldwide and extremely few from India. We present a case of 4-year-old girl who presented with multiple painless nodular masses on the scalp. On fine needle aspiration cytology, a diagnosis of benign spindle cell lesion was provided, the scalp lesions were excised and on histopathology the diagnosis of JHF was made. Retrospectively, the cytology slides were reviewed and the features were consistent with the diagnosis. A diagnosis of JHF was made based on the classical clinical features, Fine Needle Aspiration Cytology (FNAC) findings and the characteristic histopathological features along with a supportive evidence based on special stains. However, the patient had a recurrence within two years. Hence, we present this rare case with recurrence.
Juvenile hyaline fibromatosis, Chromosome 4q21 mutation, CMG2 protein, Recurrence
Case Report
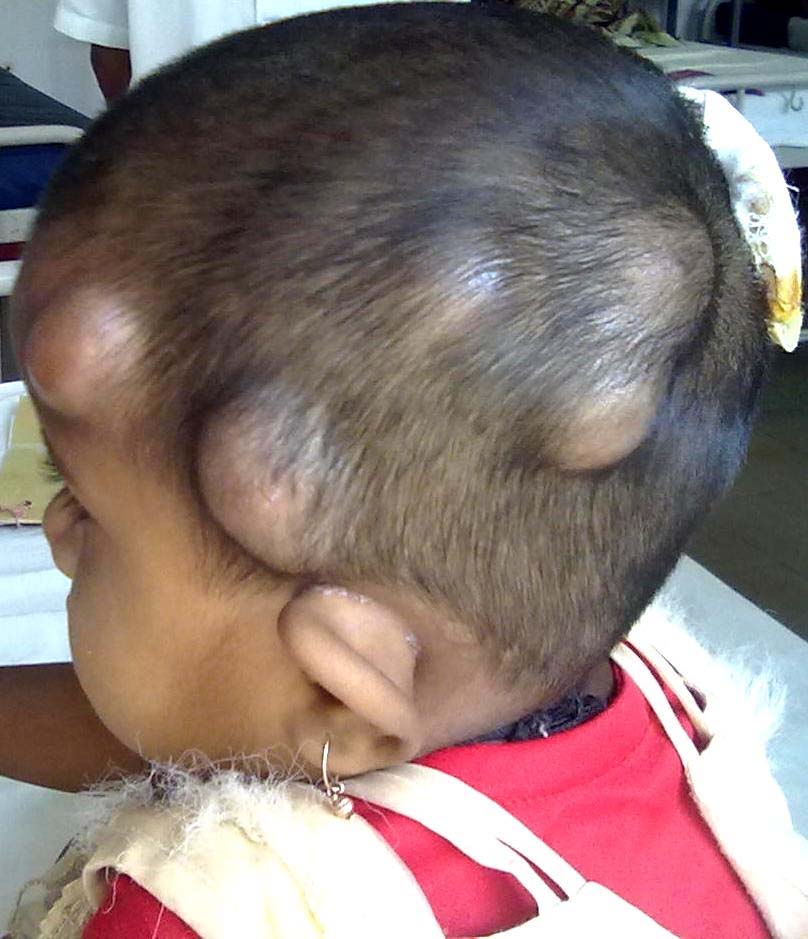
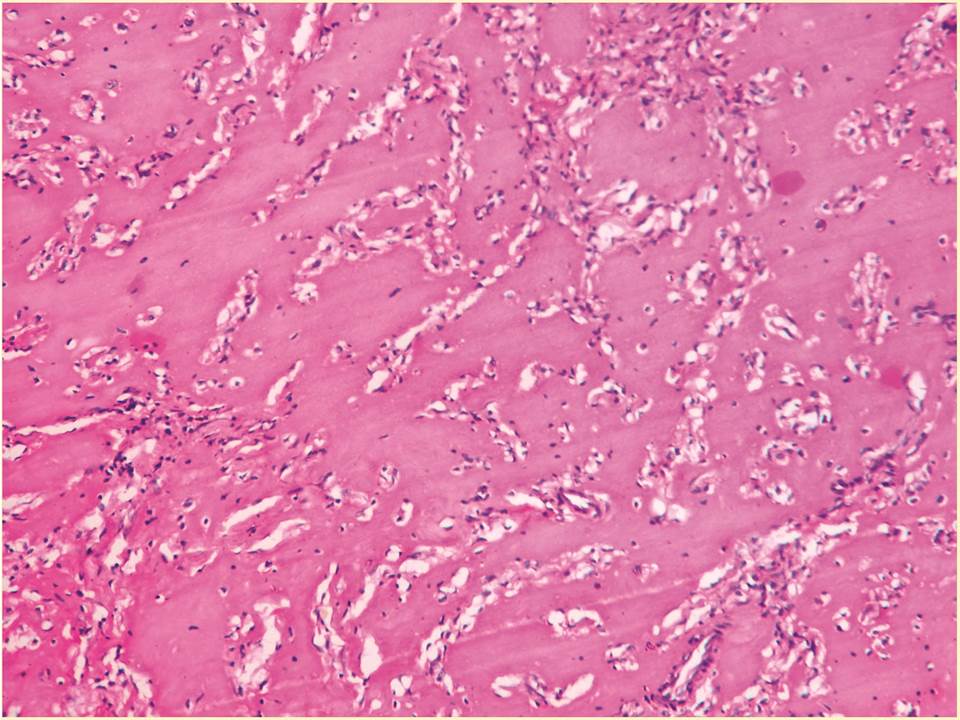
A 4-year-old girl presented with multiple painless nodular masses on the scalp, first noticed when the child was around 1-year-old [Table/Fig-1]. On examination, an anal papule also was noted. No gingival hyperplasia, osteolytic lesions or joint deformities were noted. On examination, the lesions were soft to firm in consistency and non-tender. There was no significant lymphadenopathy or hepatosplenomegaly. Hematological and biochemical investigations were within normal limits. No history of consanguinity was present in the parents. Fine-needle aspiration of the scalp mass revealed benign spindle-shaped cells with an eosinophilic ground substance in the background. A diagnosis of benign spindle cell lesion was made. The lesions were excised. Grossly, the nodules were of variable sizes, greyish-white and the cut-surface showed a gelatinous grey-white appearance. Microscopically, a poorly circumscribed lesion composed of benign oval and spindle shaped cells in groups and cords in an abundant, homogenous, eosinophilic matrix was seen. There were no mitotic figures [Table/Fig-2]. The ground substance was PAS positive and negative for toluidine blue. Based on the clinical findings, a review of cytology smears and histopathological slides, a diagnosis of JHF was made. However, the patient had recurrence of lesions on the scalp within two years.
Multiple nodular masses on the scalp
Benign oval to spindle-shaped cells arranged in cords and groups seen in an abundant, homogenous, eosinophilic matrix
Discussion
JHF is a rare hereditary disorder with variable penetrance. The disease is characterized by multiple tumourous muco-cutaneous proliferations, joint contractures, gingival hypertrophy and osteolytic lesions. Occasional patients have perianal papillomatous lesions. It is hypothesized that JHF is a connective tissue disorder with a progressive nature. This crippling disorder requires repeated surgical interventions and physiotherapy. Rehabilitation and counseling of parents play an essential role in treatment of these patients [1-4].
JHF is a rare mesenchymal dysplasia, was first described by Drescher et al., and Murray J, as “peculiar cases of molluscum fibrosum”. The other synonyms include puretic syndrome, disseminated painful fibromatosis & fibromatosis hyalinica, multiplex juvenilis [1,3].
JHF is a fatal disease with an unknown etiology caused due to mutation of capillary morphogenesis protein 2 gene. It is an inherited AR disorder caused due to mutation of CMG2 gene on chromosome 4q21. There is disruption of the formation of basement membranes, allowing the hyaline substance to leak through and build up in various body tissues like skin, joints and bones leading to the development of skin papules, gingival hyperplasia, osteolytic lesions in bone and joint contractures [5,6].
There are speculations that this disorder occurs due to impaired synthesis of pro-collagen or tropocollagen, an increased or faulty synthesis of glycoaminoglycans by fibroblasts and an association with impaired collagen type VI metabolism. Breier et al., demonstrated a significantly decreased TIIIC and increased TIC synthesis and degradation in fibroblasts cell cultures of normal human skin of a patient with JHF [7].
However, the gene for JHF has been mapped to 4q21 (name of the gene “anthrax toxin receptor 2” – ANTHRAX2; cytogenetic location: 4q21.21). Approximately, 10 mutations in ANTXR2 gene have been shown to cause JHF. It has been hypothesized that JHF is caused due to aberrant synthesis of GAGs by fibroblasts. In JHF, dermatan sulfate, chondroitin sulfate and hyaluronan with dermatan sulfate predominanace was noted in contrast to predominance of hyaluronan in normal skin [5,8].
Patients present with large cutaneous nodules especially around the head and neck regions, joint contracture, gingival hypertrophy and osteolytic lesions. Occasional reports of anal papules have been reported, as was seen in our case. The effects of JHF increases with age. Most patients have a positive family history especially on the mother’s side. One third of the affected children are siblings, often born to consanguineous marriages. There is no sex predilection [1,2,7,9].
The diagnosis is based on histopathology. However, in a study by Geethamani et al., a cytomorphological diagnosis was done for the first time in literature. The lesion can be suspected based on the history and classical clinical features. On FNA, the smears show single scattered and few groups of cells. The cells are oval to spindle shaped and have normochromic nuclei with rounded ends, no nucleoli. No mitosis is appreciated. Distinct homogenous, pale, eosinophilic material in the background of cell groups will be seen. The histopathology appearance is characteristic. Benign oval to spindle-shaped cells arranged in cords and groups are seen in an abundant, homogenous, eosinophilic matrix [6,10].
The differential diagnosis of JHF includes infantile systemic hyalinosis, neurofibromatosis, gingival fibromatosis, nodular amyloidosis, congenital generalized fibromatosis, multicentric infantile fibromatosis, lipoid proteinosis and Winchester syndrome [3].
No specific treatment is available for JHF. The treatment is only aesthetic and its aim is to limit orthopedic disability. Early tumorectomy, intralesional systemic steroids, capsulotomy, partial gingivectomy and physiotherapy to limit flexion contractures are done for these patients, but many cases have recurrences. The parents should be counselled about the progressive nature of the disease and 25% chance of development of disease in future offspring [5,6].
Conclusion
Diagnosis of JHF can be made based on clinical, cytological and histopathological findings. Early interventional and supportive treatment, rehabilitation along with counselling forms the important aspects of the treatment.
[1]. B Norman, N Soni, N Madeen, Anaesthesia and juvenile hyaline fibromatosisJ Anaes 1996 76:163-66. [Google Scholar]
[2]. FV Brandao, CMR Silva, B Gontijo, ACM Guedes, Case for diagnosisAn Bras Dermatol 2009 84(6):677-9. [Google Scholar]
[3]. GF Nakipoglu, A Dogan, M Dogan Aslan, N Ozgirgin, Juvenile hyaline fibromatosis: case presentation (the rehabilitation of three siblings)Turk J Med Sci 2004 34:67-71. [Google Scholar]
[4]. KC Nischal, D Sachdev, V Kharkar, S Mahajan, Juvenile hyaline fibromatosisJ Postgrad Med 2004 50(2):125-6. [Google Scholar]
[5]. K Raja, Z Abbas, NH Luck, SM Hassan, Three years old child with juvenile hyaline fibromatosis presenting with rectal bleedingJPMA 2013 63(3):396-8. [Google Scholar]
[6]. J Krishnamurthy, BS Dalal, Sunita MV Gubbanna, Juvenile hyaline fibromatosisInd J Dermatol 2011 56(6):731-33. [Google Scholar]
[7]. F Breier, S Fang-Kricher, K Wolff, W Jurecka, Juvenile hyaline fibromatosis : impaired collagen metabolism in human skin fibroblastsArch Dis Child 1997 77:436-40. [Google Scholar]
[8]. KR Kiran, Juvenile hyaline fibromatosis: a case report and review of literatureInt J Biol Med Res 2012 3(4):2655-7. [Google Scholar]
[9]. M Dhingra, S Amladi, S Savant, C Nayak, Juvenile hyaline fibromatosis and infantile sysytemic hyalinosis: Divergent expressions of the same genetic defect?Indian J Dermatol Venereol Leprol 2008 74:371-4. [Google Scholar]
[10]. V Geethamani, S Ravindra, VV Reddy, Fine needle aspiration cytology of juvenile hyaline fibromatosisActa Cytol 2007 51(4):624-6. [Google Scholar]