Chemically, oxidative stress is associated with an increased production of oxidizing species or a significant decrease in the effectiveness of antioxidant defenses, such as glutathione [1]. The effects of oxidative stress depend upon the size of these changes, with a cell being able to overcome small perturbations and to regain its original state. However, more severe oxidative stress can cause cell death and even moderate oxidation can trigger apoptosis, while more intense stresses may cause necrosis [2].
Production of reactive oxygen species (ROS) is a particularly destructive aspect of oxidative stress. Such species include free radicals and peroxides. Some of the less reactive of these species (such as superoxide) can be converted into more aggressive radical species by oxidoreduction reactions with transition metals or other redox cycling compounds (including quinones), that can cause extensive cellular damage [3]. The major portion of long term effects is inflicted by damage on DNA [4].
There are several cellular mechanisms that counterbalance the production of ROS, one of the most important being reduced glutathione [5].
In the setting of CAD, reactive oxygen species are proposed to play a significant role in tissue necrosis and reperfusion injury [6,7]. Increased free radicals and various inflammatory mediators in atherosclerosis can impair collagen synthesis, which is required for maintainence and repair of the fibrous cap and for triggering degradation of extracellular matrix macromolecules, which further weaken plaque’s fibrous cap, enhance its vulnerability to rupture and lead to the progression from SA through unstable angina, ultimately leading to MI. The severity and duration of imbalance between myocardial oxygen supply and demand determine as to whether the damage is reversible or permanent, with subsequent myocardial necrosis leading to MI. Ischaemia also causes characteristic changes in electrocardiogram, such as repolarization abnormalities, as is evidenced by inversion of T waves and when they are more severe, displacement of ST segments [8]. The present study was conducted to establish the role of oxidative stress in progression of CAD.
Materials and Methods
This study was a case control prospective study which was conducted in Govt. Medical College, Amritsar, India from 01/12/2008 to 01/11/2010. A total of 100 subjects were included and they were divided into two groups:
GROUP I comprised of 50 clearly defined cases of CAD, who were between 40-70 years of age, who attended the OPD or were admitted in the Department of Medicine of the institution. These patients were further divided into three groups: those with SA, unstable angina and MI, on the basis of clinical features, ECG (Electrocardiography) reports and TMT (Tread Meal Test) [9].
GROUP II comprised of 50 age and sex matched healthy individuals from the general population, who volunteered for getting included in the present study.
Informed consents were taken from all the subjects who were included in the study. The study was approved by the ethical committee of the institution. The individuals who were either taking diuretics or oral contraceptive pills were excluded from the study group. All the individuals who were selected for study were examined and investigated for lipid profile, blood reduced glutathione and serum MDA.
Blood glutathione estimation (GSH): Beutler E et al., [10].
Principle: This method is based on the development of a relatively stable yellow colour, when DTNB (5-5 dithionine 2-nitrobenzoic acid) is added to a sulphhydryl compound.
MDA: Buege and Aust [11].
Principle: Lipids, mainly PUFA, are highly susceptible to peroxidation by various oxidizing free radicals which are formed by ionizing radiations and nonenzymatic reactions. Cycloperoxides are formed as a result of these peroxide reactions, which give MDA by cleavage. MDA which is thus formed reacts with thiobarbituric acid (TBA) to form a pink coloured chromophore, which absorbs maximally at 535 nm. MDA is taken as a marker of oxidative stress and its level in serum samples is estimated by the Buege and Aust method.
Blood lipid profile: Serum Total Cholesterol [12]: Estimated by Zlatki’s method which was modified by Zak. HDL-c: Estimated by using the method of Burnstein M et al., [13]. Triglycerides: Estimated by using the method of Bucolo and David. This was done by an enzymatic method by using a triglyceride kit (Lyphozyme) [14]. Calculation of low density lipoprotein cholesterol (LDL-C): This was done by using Friedewald’s formula [15].
Statistical Analysis
All the parameters were recorded, tabulated, depicted and analyzed by using unpaired Student’s t-test and the results were depicted as not significant (NS), significant (p<0.05) and highly significant (p<0.001).
Results
[Table/Fig-1] shows the significant increase in the levels of serum lipids and lipoproteins (TC, TG ,LDL and VLDL) in the patients of CAD as compared to those in controls. There was also a significant increase of the lipid peroxidation product, MDA and reduction of antioxidant, Glutathione, in patients of CAD and in the control group. The difference in oxidative stress markers was statistically highly significant between SA and unstable angina and SA and MI, whereas the difference was not significant between USA and MI The lipid peroxidation product, MDA, showed a significant negative correlation with antioxidant, GSH, indicating the neutralization of GSH with increasing oxidative stress. It is also believed that LDL is mainly oxidized in the intima where antioxidant concentration is less.
Variations of Lipid profile and oxidative stress markers in coronary artery disease
| Parameter | Patients of CAD | NHI |
|---|
| TC (mg%) | 229.87±40.17 | 184.14±33.08 |
| TAG(mg%) | 168.70±61.03 | 113.96±22.38 |
| HDL(mg%) | 40.38±6.01 | 49.54±4.72 |
| LDL(mg%) | 155.16±35.12 | 111.34±29.17 |
| VLDL(mg%) | 33.50±12.1 | 22.78+4.47 |
| GSH (mg%) | 19.66±10.47 | 45.96±11.13 |
| MDA(mMoles/L) | 6.06±1.61 | 1.93±0.66 |
Discussion
There is now a consensus that atherosclerosis represents a state of heightened oxidative stress which is characterized by lipid and protein oxidation in the vascular wall. Oxidative stress is caused by an imbalance between the production of ROS and a biological system’s ability to readily detoxify the reactive intermediates or easily repair the resulting damage. ROS or free radicals are involved in mediation of endothelial injuries, leading to programmed cell death or apoptosis and to a form of apoptosis which is characterized by detachment of endothelial cells, which is called anoikis. ROS are known to quench NO with the formation of peroxynitrite, which is a cytotoxic oxidant and through nitration of proteins, which affect endothelial function and lead to atherosclerosis and other cardiovascular diseases [14]. With regards to atherosclerosis, vascular antioxidants need to protect against 1e-(radical) and 2e-oxidants, both within and outside cells [12,15].
ROS can be formed in the heart and other tissues by several mechanisms; they can be produced by xanthine oxidase (XO), NAD(P)H oxidases, cytochrome P450; by autooxidation of catecholamines; and by uncoupling of NO synthase (NOS) [16]. NO contains an unpaired electron, and under certain conditions, it can react with O2•– to form peroxynitrite (ONOO•–), a powerful oxidant. Angiotensin II (ATII), PDGF, and TNF-α, can also induce H2O2 and O2•– formation via activation of the NAD(P)H oxidases [17,18]. This NAD(P)H-dependent pathway has been best described in vascular smooth muscle cells, but it has also been documented in other cell types, which include cardiomyocytes [19–21].
Lipid peroxidation, for example, is a well-characterized effect of ROS that results in damage to the cell membrane, as well as to the membranes of cellular organelles [16,22].
ROS activity in the vessel wall, for example, is thought to contribute to the formation of oxidized LDL, a major contributor to the pathogenesis of atherosclerosis [23]. ROS-associated activation of MMPs may play an important role in vessel plaque rupture, initiating coronary thrombosis and occlusions [24].
There are several cellular mechanisms that counterbalance the production of ROS, which include enzymatic and nonenzymatic pathways [25]. Among the best-characterized enzymatic pathways are catalase, glutathione peroxidase, superoxide dismutases (SODs) and thioredoxin and thioredoxin reductase pathways. Nonenzymatic mechanisms include intracellular antioxidants such as the vitamins C, E, and β-carotene (a precursor to vitamin A), ubiquinone, lipoic acid, and urate [25]. They also include glutathione, which acts as a reducing substrate for the enzymatic activity of glutathione peroxidase.
The present study [Table/Fig-1] showed a significant decrease in the levels of GSH in patients of CAD as compared to those in controls, which suggested that depressed GSH levels may be associated with enhanced protective mechanisms to oxidative stress in AMI [26].
Glutathione peroxidases operate in concert with glutathione reductase, that catalyzes the reduction of GSSG at the expense of NADPH.
GSSG +NADPH + H+ → 2GSH + NADP+
Cavalca V et al., also demonstrated similar findings of significantly decreased GSH in patients of CAD as compared to that in control subjects [27].
Lipid peroxidation refers to the oxidative degradation of lipids. Catalá A, described that the general process of lipid peroxidation consisted of three stages: initiation, propagation, and termination [28].
A variety of lipid byproducts are produced as a consequence of lipid peroxidation, some of which can exert adverse and/or beneficial biological effects [29,30].
An example of a primary lipid peroxidation aldehyde is MDA. The levels of MDA are significantly higher in patients as compared to those in normal healthy individuals. This increase in MDA despite neutralization of free radicals by GSH, depicts the persisting oxidative stress and depletion of protective mechanisms, which lead to persistent damage by free radicals [31].
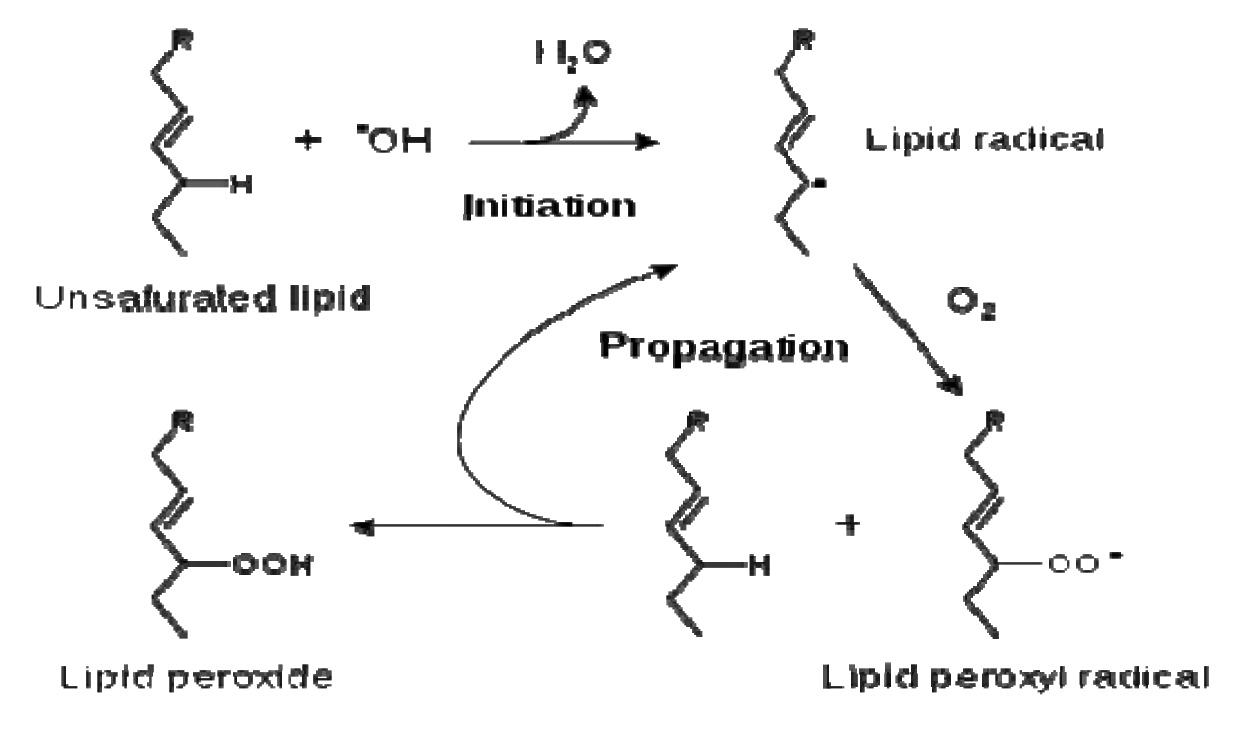
The theory which was suggested by Gotto AM et al., gives an explanation about the increased concentration of secondary products of lipid peroxides i.e. MDA in the blood of patients with atherosclerotic occlusive arterial disease, as well as about the association and the concentrations of lipid peroxides and severity of atherosclerosis. Lipid peroxides may inhibit the formation of prostacyclin (PGI2) in endothelium, resulting in platelet aggregation. Also, lipid peroxides reduce the activity of antithrombin III and accelerate the blood clotting process [32].
Kharb S showed that GSH levels were significantly decreased in patients with AMI than in the controls, (p<0.001). MDA levels were significantly elevated in AMI patients as compared to those in controls (p<0.05) [26].
The present study evaluated the oxidant – antioxidant status in subgroups of CAD. [Table/Fig-2] clearly shows that there was a difference in the levels of GSH and MDA in SA as compared to those in unstable angina and MI. But unstable angina and MI showed no such variations. According to Dubois-Randé et al., plasma MDA levels of patients who presented with unstable angina (p < 0.01) and acute MI (p < 0.05) were higher than those in patients with SA and in normal volunteers, whereas, there was no difference in these parameters between unstable angina and MI groups [33].
Variations of reduced Blood Glutathione(GSH) and Serum Malonyldialdehyde (MDA) in stable angina, unstable angina and MI
| Type of CAD | GSH (in mg%) | MDA (in mMoles/L) |
|---|
| Mean±SD | Mean±SD |
|---|
| SA n = 11 | 29.32±10.31 | t = 3.092 p<0.001 Highly Significant | 3.97±0.54 | t = 5.634 p<0.001; Highly significant |
| USA n = 16 | 16.88±10.25 | 6.28±1.27 |
| SA n = 11 | 29.32±10.31 | t = 3.811 p=0.001;Highly Significant | 3.97±0.54 | t =7.402 p<0.001;Highly significant |
| MI n = 23 | 16.99±8.05 | 6.90±1.25 |
| USA n = 11 | 16.88±10.24 | t = 0.039 p = 0.969; Not Significant | 6.27±1.27 | t = 1.533 p=0.134; Not Significant |
| MI n = 23 | 16.99±8.05 | 6.90±1.25 |
[Table/Fig-3] shows the coefficient of correlation between GSH and MDA in both patients of CAD and in normal healthy individuals. The results showed a significant negative correlation between GSH and MDA, with coefficient of correlation being -0.335 (significant) in patients of CAD and -0.27 (not significant) in normal healthy individuals. This may be due to utilization of GSH in quenching free radicals and still persisting oxidative stress, which may have caused an increase in MDA levels due to increased lipid peroxidation. Stevuljevica JK et al., observed similar results, with levels of MDA showing a significant negative correlation with GSH in patients of CAD as compared to those in normal healthy individuals, in a study which was conducted in University of Belgrade [31].
Correlation of Serum Malonyldialdehyde (MDA) with Reduced Blood Glutathione(GSH) in patients of CAD and Controls
| Group I (CAD) | Group II (NHI) |
|---|
| Mean±SD | r value | Significance | Mean±SD | r value | Significance |
|---|
| GSH | 19.66±10.47 | − 0.335 | p=0.017 Significant | 45.96±11.13 | -0.278 | p = 0.051; Not Significant |
| MDA | 6.0±1.61 | 1.93±0.66 |
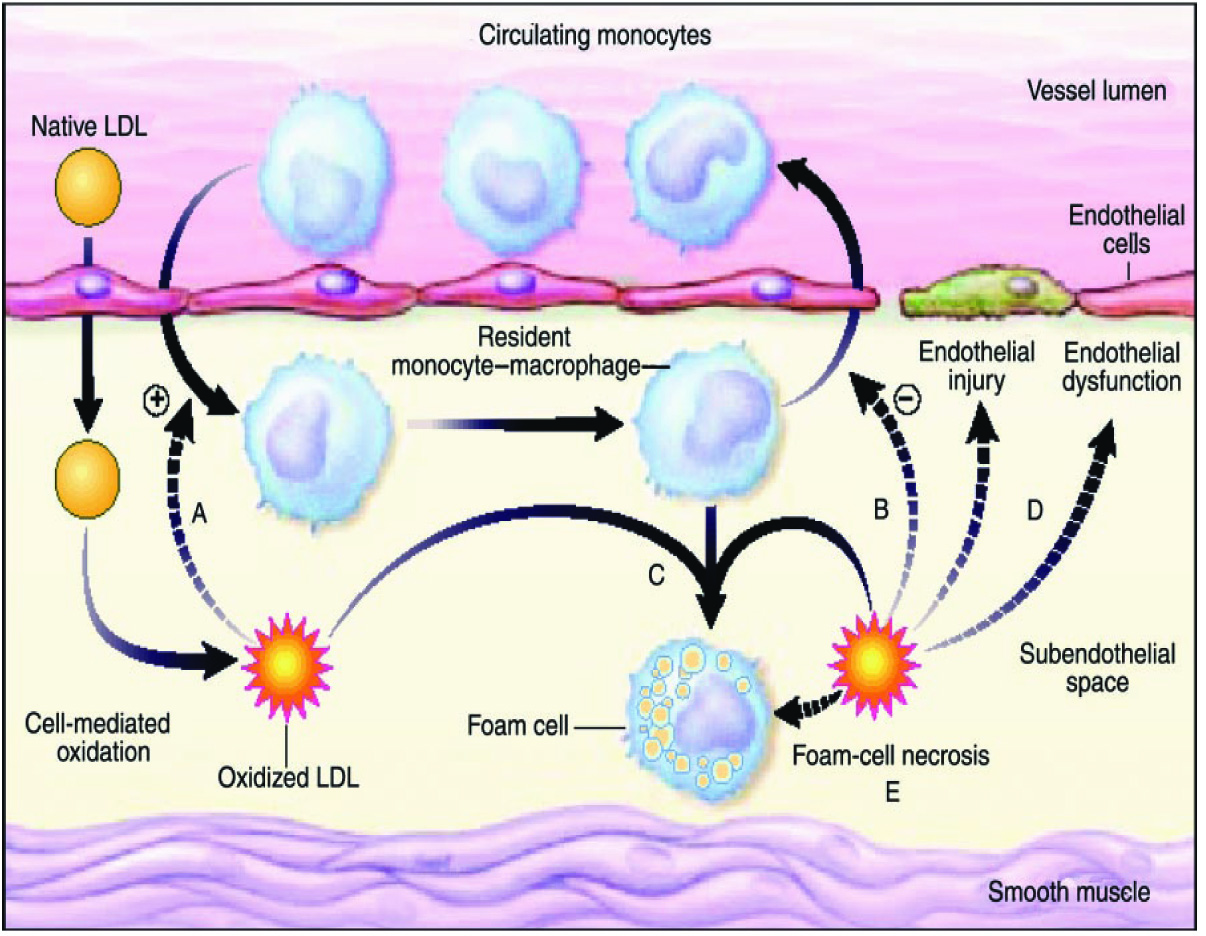
This may be due to the fact that increased free radicals and various inflammatory mediators in atherosclerosis can impair collagen synthesis which is required for maintainence and repair of the fibrous cap. During the evolution of a plaque, the free radicals and various inflammatory biomarkers trigger the egress of lipoproteins and leucocytes, causing cell death and calcification, which lead to lesion formation and which cause degradation of extracellular matrix macromolecules, which further weaken plaque’s fibrous cap, enhance its vulnerability to rupture and lead to the progression from SA through unstable angina, ultimately leading to MI [8] [Table/Fig-4].
Oxidative modification hypothesis of atherosclerosis
Limitations
1. Follow ups of the patients were not done after supplementation with antioxidants. 2. Previously diagnosed patients were included in the study. The results may be affected by the medications that they were taking.
Conclusion
The present study suggested that the significantly increased levels of lipid peroxidation marker, MDA and reduction in the antioxidant, GSH, represented state of heightened oxidative stress in atherosclerosis. Further, the increase in these markers, with an increase in the duration of the disease, enhance the vulnerability of fibrous caps of plaques to rupture and lead to progression of CAD from SA through unstable angina, leading to MI. It may be concluded that early supplementation with antioxidant vitamins may delay this progression.