Hind Brain Agenesis A Rare Imaging Findings In Cerebro Cerebellar Lissencephalic Syndrome
Praveen M. Mundaganur1, Pradeep Solwalkar2, Vishal Nimbal3
1 Assistant Professor, Department of Radiology, Bldes HospitalBijapur, Karnataka-586103, India.
2 Assistant Professor, Department of Radiology, BLDE’s B M Patil Medical College, Bijapur – Karnataka, India.
3 Senior Resident, Department of Radiology, BLDE’s B M Patil Medical College, Bijapur – Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Praveen M. Mundaganur, Department of Radiology, Bldes Hospital Bijapur, Karnataka, Pin-586103
Phone: 9916278706,
E-mail: Praveen_mdrd@Yahoo.Com
A case report of cerebro cerebellar lissencephaly shows complete agenesis of cerebellum and brainstem which is rare imaging finding of group lissencephaly (type I lissencephaly). Though agenesis of cerebellum and brainstem were included in literature, in most of the cases we saw a hypoplasia or atrophy of cerebellum in lissencephaly syndrome. The CT scan findings of this patient shows features of lissencephaly with complete agenesis of brain stem and cerebellum associated with multiple congenital abnormalities.
Hind brain agenesis, Miller-Dieker syndrome, Cerebellar lissencephalic syndrome ligament
Case Report
A newborn boy was referred for evaluation of neonatal seizures. He was born at term, to a 29 year-old g2p1 mother after an uneventful pregnancy. Both parents were healthy and unrelated. The family history was non-contributory. His condition at birth was good: apgar scores being 9/10 and 10/10 at 1 and 5 minutes respectively. He weighed 2.7 Kg at birth and the occipitofrontal circumference measured 33 cm (10th - 25th percentile). On the second day of life, he was noted to have several episodes of twitching of the face and jerky movements of both upper limbs. He was thus referred for further management.
On admission to our institution, no dysmorphic features were noted and the rest of the physical examination including the neurological examination and fundoscopy did not reveal any abnormality. The complete blood count, haemogram and serum electrolyte levels were normal, random blood glucose was 3.8 Mmol/l. Results of cerebrospinal fluid analysis was normal. CSF and blood cultures were sterile. Titres of serum antibodies to toxoplasma, CMV, herpes and rubella were not elevated. EEG showed frequent sharp waves and spikes of high voltages over the frontal and temporal regions bilaterally.
Subsequently CT scan of the brain revealed dilatation of both lateral ventricles and suprasellar cisterns with agenesis of the brain stem and cerebellum. The brain surface was relatively smooth suggestive of deficiency in sulci and gyri. A diagnosis of cerebrocerebellar lissencephaly was made. Chromosome study was a 46xy karyotype. Phenobarbitone (5mg/kgl day) as a single dose was commenced and fits were under control. He was discharged on day ten of life. His neurodevelopment was markedly delayed and his seizures recurred but these were controlled by increasing the dose of the anticonvulsant.
Discussion
Traditionally, cases of lissencephaly have been divided into two groups based on clinicopathologic type. In type I or classic lissencephaly, the normal six-layer cortex seen at histologic analysis is replaced by an abnormally thick, remodeled, four-layer cortex. Classic lissencephaly may result from an isolated lissencephaly sequence or may be associated with Miller-Dieker syndrome or Norman-Roberts syndrome. Dobyns et al., [1] described a grading system for type I lissencephaly, with degrees of severity ranging from diffuse agyria to mixed agyria and pachygyria, pachygyria only, and subcortical band heterotopia. Clinically, Miller-Dieker syndrome can be differentiated from isolated lissencephaly sequence by the presence of facial dysmorphism and a more severe grade of lissencephaly. Miller-dieker syndrome is the form of lissencephaly most easily recognized on prenatal images because of the striking and total lack of sulci. Other forms may escape prenatal detection because milder or lesser degrees of sulcal developmental arrest may occur after the primary sulci have formed.
Type II lissencephaly is pathologically distinct from type I and, at histologic analysis, is characterized by a disorganized unlayered cortex. While in type I lissencephaly many neurons fail to reach the cortical plate, in type II lissencephaly many neurons move too far into the subpial space [2–4]. Type II lissencephaly, also known as cobblestone complex, is observed in some forms of congenital muscular dystrophy that are associated with cortical mal development, including Walker-Warburg (also known as harde [hydrocephalus, agyria, and retinal dysplasia with or without encephalocele]) syndrome, muscle-eye-brain disease, and Fukuyama-Type congenital muscular dystrophy. The diagnostic criteria for Walker-Warburg syndrome as described by Dobyns et al., [5] include type II lissencephaly, cerebellar malformation, retinal malformation, and congenital muscular dystrophy. Brainstem abnormalities also may be present. They are difficult to detect at fetal ultrasound, but MRI images demonstrate specific findings.
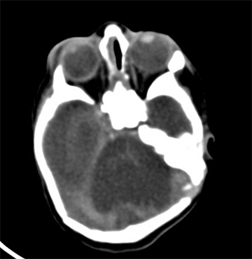
Shows agenesis of cerebellum and brain stem with small posterior fossa.
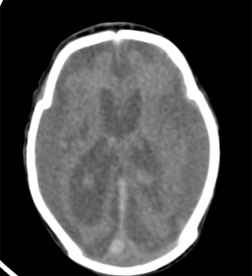
shows smooth brain surface with paucity of gyri and sulci; atrophic brain and dilated lateral ventricles.
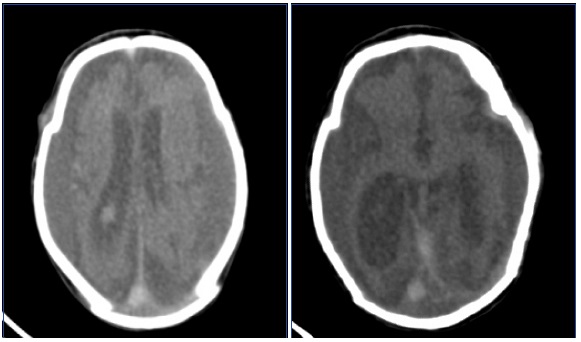
shows smooth brain surface with paucity of gyri and sulci; atrophic brain and dilated lateral ventricles. There is agenesis/atrophy of carpus callosum.
In a more recent classification system proposed by Barkovich et al., malformations due to abnormal neuronal migration are categorized in three main groups [6]. Group A, also known as the lissencephaly–subcortical band heterotopia spectrum, includes type I or classic lissencephaly, as well as lissencephaly with agenesis of the corpus callosum, lissencephaly with cerebellar hypoplasia, and lissencephaly not yet classified. Group B, also known as cobblestone complex, includes type II lissencephaly. Group C comprises heterotopias other than subcortical band heterotopia [6].
Reelin is an extracellular matrix-associated protein important in the regulation of neuronal migration during cerebral cortical development. Point mutations in the reln gene have been shown to cause an autosomal recessive human brain malformation termed lissencephaly with cerebellar hypoplasia (lch). Recent work has raised the possibility that reelin may also play a pathogenic role in other neuropsychiatric disorders [7].
Conclusion
Complete agenesis of hind brain in lissencephaly is also seen as spectrum of findings even though hypoplasia is common .Although it requires detailed clinical,imaging and genetic correlation in the diagnosis of lissencephaly syndrome and classification, imaging plays major role. MRI is the modality of choice to fully characterise the abnormalities
[1]. Dobyns WB, Truwit CL, Ross ME, Differences in the gyral pattern distinguish chromosome 17-linked and X-linked lissencephalyNeurology 1999 53:270-77. [Google Scholar]
[2]. Haltia M, Leivo I, Somer H, Muscle-eye-brain disease: a neuropathological studyAnn Neurol 1997 41:173-80. [Google Scholar]
[3]. Miller G, Ladda RL, Towfighi J, Cerebroocular dysplasia–muscular dystrophy (Walker Warburg) syndrome: findings in 20-week-old fetusActa Neuropathol (Berlin) 1991 82:234-38. [Google Scholar]
[4]. Squier MV, Development of the cortical dysplasia of type II lissencephalyNeuropathol Appl Neurobiol 1993 19:209-13. [Google Scholar]
[5]. Dobyns WB, Pagon RA, Armstrong D, Diagnostic criteria for Walker-Warburg syndromeAm J Med Genet 1989 32:195-210. [Google Scholar]
[6]. Barkovich AJ, Kuzniecky RI, Jackson GD, Guerrini R, Dobyns WB, Classification system for malformations of cortical development: update 2001Neurology 2001 57:2168-2178. [Google Scholar]
[7]. Am J Med Genet B Neuropsychiatr Genet 2007 Jan 5 144B(1):58-63. [Google Scholar]