A Case of Diamond Blackfan Anemia (DBA) with Mutation in Ribosomal Protein S19
John Solomon1, Rugmini Kamalammal2, Godfred Antony Menezes3, Mohamed Yaseen Sait4, Harita Lohith5, Revathy Ramalingam6
1 Professor & Head, Department of Paediatrics, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
2 Associate Professor, Department of Paediatrics, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
3 Scientist & Incharge, Central Research Laboratory- CRL, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
4 Postgraduate student, Department of Paediatrics, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
5 Postgraduate student, Department of Paediatrics, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
6 Research Assistant, Central Research Laboratory- CRL, Sree Balaji Medical College & Hospital (Bharath University), Chromepet, Chennai, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. P. John Solomon, Professor, Department of Paediatrics, Paediatric Haematology and Oncology and H.O.D., Department of Paediatrics., Sree Balaji Medical College & Hospital, Chennai-600044, India.
Phone: +91 9444610092,
E-mail: pjohnsolomon@yahoo.co.in
Diamond Blackfan Anemia (DBA) is a rare disorder which presents with anemia in early infancy. This disorder is genetically and clinically heterogeneous in nature. The inheritance is mainly autosomal dominant. Approximately 25% of the cases are associated with craniofacial anomalies and some cases may end up in malignancy. The diagnosis is made by blood investigations, and bone marrow studies in which red cell precursors are reduced or absent. Screening for the mutations including those encoding for ribosomal proteins in the patient and the family members confirms the diagnosis. Human Leukocyte Antigen (HLA) matched hemopoietic stem cell transplantation is the treatment of choice. In other cases, corticosteroids and cyclosporine A have been tried. The haemoglobin level is maintained with packed red cell transfusion. We are presenting here a female baby who had anemia at birth and was brought to us at the age of 2 months. The diagnosis of DBA was made since the patient presented with anemia and showed reticulocytopenia, gross reduction in Red Blood Cell (RBC) count, and reduction in red cell precursors in the bone marrow. Genetic screening revealed mutation in ribosomal protein S19 (RPS19) gene in both the infant and the father.
Anemia, Mutations, Human Leukocyte Antigen
Case Report
A 2-month-old female baby born of a third degree consanguineous marriage was brought to our hospital with history of being pale at birth and having received 2 blood transfusions, one at 48 hours of life and the second transfusion within one month of life. There was no history of jaundice. The developmental milestones were appropriate for age.
The infant’s elder sibling also presented with anemia in another hospital at four months of age and was diagnosed as DBA. He was treated with blood transfusions and finally he died at the age of 2 years.
On examination she was severely anemic but there was no icterus. The baby weighed 3.5kg and the head circumference was 31 cm against the expected weight of 3.8 kg and head circumference of 37-39 cm. Anterior fontanelle was very small and posterior fontanelle was closed. The occipital region was flat. Examination of other systems did not reveal any abnormalities.
The haemoglobin level was 2.6gm/dl and hematocrit was 8.3%. The blood counts showed reduction in both RBC count (0.83million/cmm) and the reticulocyte count (0.2%). The White Blood Cell (WBC) count and platelet count were normal. The red cells were normocytic and HbF was within normal limits. These two normal findings can be attributed to the patient receiving two blood transfusions before coming to our institution, and because of the same reason we could not perform erythrocyte adenosine deaminase (eADA) estimation. Total serum bilirubin level was 0.6mgm/dl.
Bone marrow aspiration showed normocellular marrow with M: E (myeloid-to-erythroid ratio) of 58:1. Red cell precursors were grossly reduced. The other cell lines were normal. Ultrasound abdomen, Ultrasound cranium, Echocardiogram and Magnetic Resonance Imaging (MRI) of the brain did not reveal any abnormality.
Genetic studies were performed for screening the mutations in RPS19 gene of proband and also of parents. The genetic analysis was carried out in the Central Research Laboratory (CRL) of our Institute after obtaining the informed consent of the parents. The genomic DNA was extracted manually by phenol chloroform isoamyl-alcohol method. Polymerase Chain Reaction (PCR) was performed using primer pairs RP13/15, RP3/4, and RP19/20 (used to amplify exons 2–3, exons 4–5, and exon 6 respectively) and PCR conditions were as per Matsson et al., [1]. The PCR products were purified, sequenced and analysed for mutations. Frame shift mutations were identified in the exon 6 of RPS19 gene of infant and also father, although father did not have any signs and symptoms of the disorder. Further, no mutations were found in the mother [Table/Fig-1].
Schematic view of the nucleotide sequence alignment of the proband and her parents against the reference sequence. (NC 000019.9, GI: 224589810). Note: Frameshift mutations found in the exon 6 of RPSI9 gene in the proband and the father have been underlined
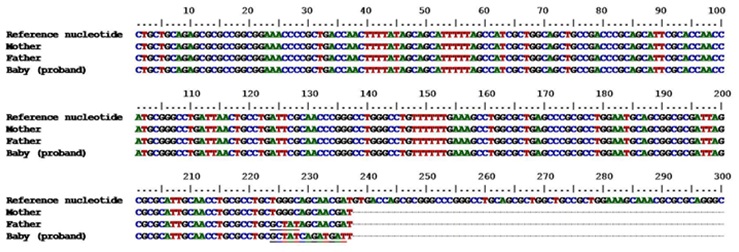
We made a diagnosis of “classical” DBA, since the baby presented with anemia at birth, reticulocytopenia, normal white cell count and platelet counts, normal marrow cellularity with paucity of red cell precursors, supporting criteria of gene mutation in RPS19 gene and positive family history. In addition, the baby had microcephaly and flat occiput. There was no evidence of any other bone marrow failure syndrome.
Since, the patient is not having another sibling, and also cannot afford the costs involved, we could not consider the possibility of hemopoietic stem cell transplantation. The patient was started on prednisolone therapy and is receiving packed red cell transfusion for the past 8 months. Based on these facts the parents were counseled.
Discussion
DBA is a rare genetic disorder distinguished by proapoptotic hematopoiesis and congenital anomalies involving craniofacial region, eyes, neck, thumb, urogenital tract, heart, and musculoskeletal structures. Some cases may present with short stature, growth retardation and learning difficulties [2]. It usually presents in infants, although presentation can vary from severe fetal anemia requiring transfusion to anemia detected as late as 6 years of age. Most cases are sporadic with a dominant or, rarely recessive pattern. An inheritance rate of 10% has been observed in patients [3]. Among the genes 25% of patients carry a mutation in RPS19 gene locus on chromosome19q13.2. Another locus has been detected on chromosome 8p (5), whereas mutations in RPS24, RPS17, RPL35A, RPL11 and RPL5 have been reported rarely [2,4]. Among RPS19 mutations, whole gene deletions, truncating mutations (nonsense or frame shift) and translocations have been observed, suggesting that haploinsufficiency is the basis of DBA pathology. A relation between ribosomal functions and erythroid aplasia is evident in DBA, but its etiology is not clear [4]. The mutations in RPL11 are more often associated with physical malformations than RPS19 mutations. The mutations in RPL5 appear to cause malformations in the heart and craniofacial region in contrast to mutations in RPS19 [5,6]. Hence, very minimal malformations in the subject prompted us to screen mutations in RPS19 gene. To the best of our knowledge though some cases of DBA have been reported in India, no genetic studies have been done in those cases [7]. In our case the mutation was found only in the exon 6 of RPS19 gene. Similarly, in a study from Italy involving patients from the DBA registry, one of the cases revealed mutation only in the exon 6 of RPS19 [8].
The differential diagnosis in our case (anemia at birth) is haemolytic disease of new born (HDN) but investigations done at the hospital where transfusion was given first has not revealed any evidence of isoimmune hemolytic anemia. Transient Erythroblastopenia of Childhood (TEC) which is another differential diagnosis for DBA presents usually after 6 months of age and is an acquired condition.
HLA matched haemopoietic stem cell transplantation offers the best hope for life. If a matched donor is not available, corticosteroid remains the main stay of treatment in DBA. The mechanism of action is not clear but approximately 70% of patients are said to respond to steroid. Increase in haemoglobin is usually seen within 2-4 weeks. Interleukins, cyclosporine, colony stimulating factors, androgens, danazol, 6-mercaptopurine, cyclophosphamide, antilymphocyte globulin, vincristine etc. have been tried in some cases with limited success [2,9]. Some will succumb to complications of treatment. A few will develop malignancy like leukemia and lymphoma at a later date.
In our case, the parents needed advice whether to have the next child or not. If classical DBA is present in the parent and offspring, or in two or more siblings the recurrence risk is upto 50%. If next pregnancy ends up in a normal sibling with matched HLA, he or she can act as a donor for haemopoietic stem cell transplantation for this patient. Antenatally, one can go in for amniocentesis or chorionic villous biopsy and select a fetus with no detectable mutation. The other alternative is to go in for intra-uterine insemination using sperm from another normal male adult. More recently Pre-implantation Genetic Diagnosis (PGD) has become an option to greatly reduce the risk of the second affected child. If mutation analyses (50% probability) and HLA typing (25% probability) are combined, the maximal success rate is only about 12.5% of embryos [2].
Conclusion
To the best of our knowledge, this is the first classical case of DBA with RPS19 mutation confirmed by genetic study in India. The case had physical abnormalities associated with RPS19 mutation as reported from other parts of the world. Further, the child has not shown any satisfactory response to corticosteroid treatment.
[1]. Matsson H, Klar J, Draptchinskaia N, Gustavsson P, Carlsson B, Bowers D, Truncating ribosomal protein S19 mutations and variable clinical expression in Diamond-Blackfan anemiaHum Genet 1999 105(5):496-500. [Google Scholar]
[2]. Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghiet U, Diagnosing and treating Diamond Blackfan anemia: results of an international clinical consensus conferenceBr J Haematol 2008 142(6):859-76. [Google Scholar]
[3]. El-Beshlawy A, Ibrahim IY, Rizk S, Eid K, Study of 22 Egyptian patients with Diamond-Blackfan anemia, corticosteroids, and cyclosporin therapy resultsPaediatrics 2002 110(4):e44 [Google Scholar]
[4]. Campagnoli MF, Ramenghi U, Armiraglio M, Quarello P, Garelli E, Carando A, RPS19 mutations in patients with Diamond-Blackfan anemiaHum Mutat 2008 29(7):911-20. [Google Scholar]
[5]. Cmejla R, Cmejlova J, Handrkova H, Petrak J, Petrtylova K, Mihal V, Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemiaHum Mutat 2009 30(3):321-27. [Google Scholar]
[6]. Gazda HT, Sheen MR, Vlachos A, Choesmel V, O’Donohue MF, Schneider H, Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patientsAm J Hum Genet 2008 83(6):769-80. [Google Scholar]
[7]. Manglani M, Lokeshwar MR, Sharma R, Diamond-Blackfan anemia: report of 6 casesIndian Paediatr 2003 40(4):355-58. [Google Scholar]
[8]. Quarello P, Garelli E, Brusco A, Carando A, Mancini C, Pappi P, High frequency of ribosomal protein gene deletions in Italian Diamond-Blackfan anemia patients detected by multiplex ligation-dependent probe amplification assayHaematologica 2012 97(12):1813-17. [Google Scholar]
[9]. Dunbar CE, Smith DA, Kimball J, Garrison L, Treatment of Diamond-Blackfan anemia with hematopoietic growth factors, granulocyte-macrophage colony stimulating factor and interleukin 3: sustained remissions following IL-3Br J Haematol 1991 79:316-321. [Google Scholar]