‘Eosinophilia’ can occur due to a large number of allergic, infectious, neoplastic, and idiopathic diseases. It can range in severity from a self-limiting condition to a life-threatening disorder. The term ‘hypereosinophilia’ refers to eosinophil levels >1500/μL, and regardless of the underlying cause can be associated with tissue and organ damage. ‘Hypereosinophilic syndrome (HES)’ is a rare disorder with sustained eosinophilia and multi-organ dysfunction in the absence of a discernable secondary cause. ‘Undefined Hypereosinophilic Syndrome’ is the most common type of primary hypereosinophilic diseases and we are reporting here one such case who presented with acute multiple embolic strokes secondary to biventricular apical thrombi and multi-organ dysfunction of a fulminant nature. This case highlights the limitation in current diagnostic criteria for HES and emphasizes the need for early intervention.
Introduction
Eosinophilia is a common disorder and may be secondary or idiopathic. ‘Hypereosinophilic Syndrome’ (HES) refers to a group of disorders characterized by sustained overproduction of eosinophils, in which eosinophilic infiltration and mediator release causes damage to multiple organs in the absence of a discernable secondary cause [1]. Neurological involvement can take the form of encephalopathy, brain infarction and sensory polyneuropathy. We are reporting here about a patient who presented to us with multiple strokes of embolic origin, but was found to have hypereosinophilia with multi-system involvement affecting the, heart, lungs and kidneys besides the CNS.
Case Report
A 68-years-old non-hypertensive non-diabetic male presented to us with a 10 days history of step wise progressive neurological deficit. For about 4 days prior to the onset of his neurological symptoms he had complained of intermittent retro-sternal chest pain and dyspnoea. Subsequently, he first developed a sudden onset predominantly distal weakness and clumsiness of the left upper limb followed 3 days later by difficulty in walking due to weakness of the left lower limb. Two days prior to admission he also developed a sudden onset weakness of the right lower limb with change in behaviour and decreased spontaneous speech. Five months back he had been diagnosed as a case of Coronary Artery Disease (CAD) when his coronary angiography had revealed 70% ostial stenosis in the obtuse marginal branch of the Left Circumflex artery and he was on medical management for the same.
On examination at the time of admission, his pulse was regular, 92/minute, and blood pressure was on the lower side (92/66mm Hg). On general examination there was a mild pallor and mild pedal oedema but no lymphadenopathy. CVS and abdominal examination were normal but there were bilateral scattered crepitations on chest auscultation. Patient was conscious and responsive to verbal commands but did not establish any eye contact and was unaware of his deficit. He denied any weakness in the limbs or any difficulty in seeing but was unable to describe or name any object placed in front of his eyes and would either confabulate or remain quiet. Blink response to threatening stimuli was absent and in spite of this objective visual loss, he would claim that he was able to see everything. Higher mental functions could not be assessed as the patient was not cooperative but ocular movements, pupillary reaction and fundus examination were normal. There was no evidence of any other cranial nerve involvement. On motor examination, formal assessment of power was not possible but the patient was moving only his right upper limb and there was minimal movement of the right lower limb and left half of the body on painful stimulus. Tone was increased in both lower limbs with brisk deep tendon reflexes and bilateral ankle clonus. Both the plantars were extensor. On the basis of history and examination a clinical diagnosis of multi-focal cerebral dysfunction with bilateral pyramidal signs and cortical blindness was entertained. Over the next 24 hours, he worsened and became drowsy, incontinent and unresponsive with a clinical picture suggestive of encephalopathy.
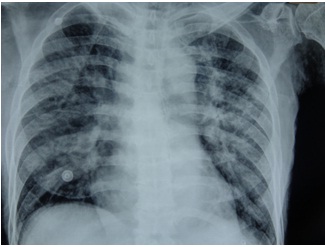
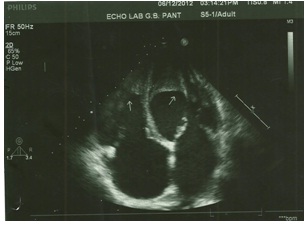
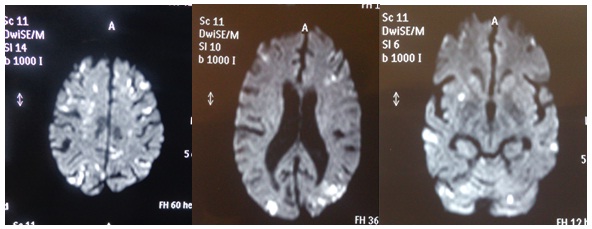
On investigations, haemogram revealed haemoglobin of 10gm % with a high total leukocyte count of 51,500 cells/ml. The differential count was also abnormal (P-12%, L-5%, M-1%, E-82%) with a high absolute eosinophil count of 42,500/μL. His peripheral blood smear showed mild anisocytosis, mild to moderate hypochromia with marked eosinophilia but no atypical cells. LFT and serum electrolytes were normal but KFT was mildly deranged with a blood urea of 76mg/dl and serum creatinine of 1.6mg/dl. Serum LDH was 427 IU/L. X-Ray chest revealed widespread, bilateral, multiple small to medium sized fluffy pulmonary infiltrates [Table/Fig-1]. On ultrasound abdomen, there was no hepato-spleenomegaly but there was dilatation of the Inferior Vena Cava (IVC) and hepatic veins. ECG showed T wave inversion in inferolateral leads. Echocardiography revealed a moderate mitral regurgitation with bi-atrial enlargement and apical clots in both the right and left ventricles. The ejection fraction was 50% and IVC was dilated and congested [Table/Fig-2]. MRI brain revealed bilateral, widespread, multiple, small areas of diffusion restriction in both the anterior and posterior circulation territories suggestive of multiple embolic infarcts [Table/Fig-3]. Further investigations were done to ascertain the cause of eosinophilia were also negative. Stool examination for parasites and serum filarial antigen were negative. Serum IgE levels were mildly raised to 446u/l. ANA, Rheumatoid factor, c-ANCA and p-ANCA were also negative. Bone marrow aspirate revealed a cellular smear with a predominance of eosinophilic precursors which comprised 50% of all the nucleated cells with no evidence of myelodyspalsia, hemoparasites or atypical cells.
X-Ray Chest showing bilateral fluffy pulmonary infiltrates
Transthoracic Echocardiography showing bi-atrial enlargement with apical thrombi in both the left and right ventricles
Diffusion weighted MR images showing multiple embolic infarcts in both the anterior and posterior circulation territories
A final diagnosis of ‘Undefined or Idiopathic Hypereosinophilic Syndrome (HES)’ related multi-organ dysfunction with neurological, cardiac, pulmonary and renal involvement was made. The cause of eosinophilia remained undefined.
In view of idiopathic hypereosinophilia and deteriorating sensorium, patient was started on steroids (IV Methyl-prednisolone, 1gm daily for 5 days followed by oral prednisolone in a dose of 60mg/day), albendazole (400mg single dose), and diethylcarbamazine (200mg three times a day, for 21 days) along with anticoagulation (subcutaneous enoxaparine-60 units twice a day). Within 3 weeks of starting therapy, AEC fell down from 42,500 to 15,000/μL, and there was a significant resolution of the pulmonary infiltrates. Although there was an initial improvement in his sensorium there was no recovery of neurological function and cardiac and renal functions remained deranged. Over the next 2 weeks, his cardiac function deteriorated leading to congestive cardiac failure, acute circulatory collapse and subsequent death.
Discussion
Eosinophilia is arbitrarily classified as mild (351 -1500 cells /μL), moderate (>1500 - 5000 cells /μL), or severe (>5000 cells /μL) [1]. In patients with hypereosinophilia of >1500/μL, eosinophils can accumulate in multiple organs including the heart, CNS and lungs, as was the case with our patient.
The most common cause of eosinophilia in industrialized nations is atopic disease but worldwide helminthic infections are the most common cause [2]. Hypereosinophilic Diseases Working Group has established 6 separate categories of primary hypereosinophilic diseases [3]. In the absence of an identifiable cause of moderate-to-severe eosinophilia and in the presence of end-organ involvement, the diagnosis of ‘Idiopathic HES’ should be considered.
HES is a leukoproliferative disorder originally described in 1975 and currently identified by three defining features: blood eosinophilia of ≥1500/μL present for more than six months; no other apparent aetiologies for eosinophilia, such as parasitic infection or allergic disease; signs and/or symptoms of eosinophil-mediated end-organ dysfunction [4]. However this definition has some inherent problems. For example as in our case, some patients may report and require therapeutic intervention well before the six months period specified in the first criterion. Once secondary causes of hypereosinophilia have been excluded, a diagnosis of ‘idiopathic HES’ can be entertained and there is no reason to withhold treatment in a patient with sustained and potentially life-threatening hypereosinophilia.
Patients may present with various combinations of symptoms and signs of end-organ damage mediated by eosinophils. In most patients, the onset of symptoms is insidious and eosinophilia is detected incidentally. However occasionally, as in our case the initial manifestations can be severe and life-threatening due to the rapid evolution of cardiac and or neurologic complications. In our case secondary causes of eosinophilia were ruled out by appropriate tests. The T-cell lymphocytic variant of HES (L-HES) has prominent skin findings (including plaques, erythroderma, and urticaria) which were lacking in our patient5. Myeloproliferative variants were ruled out by a normal serum B12 and the absence of anaemia - thrombocytopenia, hepato-spleenomegaly and circulating leukocyte precursors [5]. In patients with ‘Undefined HES’, total leukocyte counts are usually less than 25,000/ml, but rarely extremely high leukocyte counts (>90,000/ml) may be seen and were associated with worse prognosis. Our patient also had a very high initial total leukocyte count with rapid onset multi-organ dysfunction and poor response to treatment.
Clinically, HES is a heterogenous disease with varied manifestations. Patients may present with non-specific symptoms such as fatigue (26%), myalgias (14%), cough (24%), low grade fever (12%), rash (12%) and dyspnea (16%) [6]. Cardiovascular lesions pathologically described as eosinophilic myocarditis or Loffler’s fibroblastic endocarditis is a major cause of morbidity and mortality in 54-73% of patients [6,7]. Eosinophil-mediated heart damage evolves through three stages: a clinically silent acute necrotic stage; an intermediate thrombotic stage and finally a fibrotic stage [6]. Cardiopulmonary symptoms and signs develop during the thrombotic and fibrotic stages. Common findings include dyspnea, chest pain, signs of left and/or right ventricular failure, mitral or tricuspid regurgitation, cardiomegaly, and T wave inversions on the electrocardiogram. Echocardiography and cardiac MRI may demonstrate intracardiac thrombi. Finally, in the fibrotic stage, progressive scarring develops resulting in endomyocardial fibrosis producing a restrictive cardiomyopathy [8]. Our patient too had symptoms of dyspnea, chest pain and T wave inversions on ECG which was initially misinterpreted as CAD. The bilateral ventricular apical thrombi and restrictive cardiomyopathy were responsible for causing multiple embolic strokes and CHF respectively.
Neurological complications in HES may be of three types: brain infarction, encephalopathy and sensory polyneuropathy [9]. Brain infarcts are due to either thromboembolism from Endomyocardial Fibrosis (EMF) or to vascular endothelial toxicity of eosinophilic cells. Patients can experience embolic strokes or transient ischemic episodes, either of which can be multiple and recurrent as in our case. Such thromboembolic episodes can be the presenting manifestation of HES. In early HES, strokes are small and occur in the arterial border zones but later larger cortical and subcortical areas also become involved. It is also important to note that although anticoagulation and antiplatelet agents are usually instituted, recurrences of emboli in adequately anticoagulated HES patients are not infrequent [9]. The second type of neurological dysfunction classically described in patients with HES is encephalopathy. Patients exhibit changes caused by a distinct encephalopathy, including changes in behavior, confusion, ataxia, and memory loss, and exhibit upper motor neuron signs with increased muscle tone, deep tendon reflexes, and a positive Babinski sign. The exact pathogenesis of encephalopathy is not known but encephalopathic changes are more likely with markedly elevated AEC. Pathological studies in cases of eosinophilia-related encephalopathy have revealed cerebral infarctions in the arterial watershed zones. It therefore seems more likely that encephalopathy in cases of HES is caused by multiple small infarcts in the arterial border zones. The advances in neuroimaging have made it possible to identify these small infarcts which were earlier missed. Though our patient initially presented with a history suggestive of multiple recurrent strokes (bi-pyramidal signs, cortical blindness, behavioural change) within a short time span of 10 days, he subsequently developed an encephalopathy like clinical picture and diffusion weighted MRI images revealed multiple small infarcts in the anterior as well as posterior circulation territories. The third type of neurologic dysfunction described in cases with HES is peripheral neuropathies which can occur in about half of HES patients with neurologic manifestations [9].
Treatment of HES is complicated and depends on the type of HES. The goal of therapy is to reduce the signs and symptoms and maintain the eosinophil count below 1500/μL. Glucocorticoids (Prednisone at doses of 1 mg/kg daily for 1-2 weeks) appear to be highly effective in treating most patients with HES [10]. In cases with extremely elevated AEC (>100,000 cells/μL), higher doses of methylprednisolone can be administered to rapidly reduce the hypercellularity. The AEC may drop dramatically in response to steroids, as was observed in our case also. Partial or complete remission can be achieved in 85 % cases at one month. Our case however, despite an initial response to steroids succumbed due to cardiac complications. A corticosteroid-sparing agent such as hydroxyurea, interferon alpha, or anti-IL-5 is indicated in the presence of significant side effects or prednisone dose exceeding 10 mg/day.
Conclusion
HES is a heterogenous disease with varied manifestations. Present case highlights that HES can be a cause of multiple embolic infarcts with a stroke or encephalopathy like presentation that can be accompanied by a rapidly progressive life threatening multi-organ dysfunction. In cases with undefined/idiopathic HES, a high AEC can provide an initial diagnostic clue and early treatment can be initiated in patients with rapidly progressive disease after ruling out secondary causes.
[1]. Roufosse F, Weller PF, Practical approach to the patient with hypereosinophiliaJ Allergy Clin Immunol 2010 126:39 [Google Scholar]
[2]. Rothenberg ME, EosinophiliaN Engl J Med 1998 May 28 338(22):1592-600. [Google Scholar]
[3]. Klion AD, Bochner BS, Gleich GJ, Approaches to the treatment of hypereosinophilic syndromes: a workshop summary reportJ Allergy Clin Immunol 2006 117:1292 [Google Scholar]
[4]. Chusid MJ, Dale DC, West BC, Wolff SM, The hypereosinophilic syndrome: analysis of fourteen cases with review of the literatureMedicine (Baltimore) 1975 54:1 [Google Scholar]
[5]. Simon HU, Rothenberg ME, Bochner BS, Refining the definition of hypereosinophilic syndromeJ Allergy Clin Immunol 2010 126:45 [Google Scholar]
[6]. Weller PF, Bubley GJ, The idiopathic hypereosinophilic syndromeBlood 1994 83:2759 [Google Scholar]
[7]. Chew CY, Ziady GM, Raphael MJ, Nellen M, Oakley CM, Primary restrictive cardiomyopathy. Non-tropical endomyocardial fibrosis and hypereosinophilic heart diseaseBr Heart J 1977 39:399 [Google Scholar]
[8]. Brockington IF, Olsen EGJ, Loffler’s endocarditis and Davies’ endomyocardial fibrosisAm Heart J 1973 85:308 [Google Scholar]
[9]. Moore PM, Harley JB, Fauci AS, Neurologic dysfunction in the idiopathic hypereosinophilic syndromeAnn Intern Med 1985 102:109 [Google Scholar]
[10]. Ogbogu PU, Bochner BS, Butterfield JH, Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapyJ Allergy Clin Immunol 2009 124:1319 [Google Scholar]