Meckel Gruber Syndrome, Encephalocele, Polydactyly, Consanguinity
Dear Sir,
We are reporting a case of Meckel-Gruber Syndrome (MGS), which was identified by its typical structural anomalies and supported by a history of consanguinity.
MGS, which was originally described by Meckel in 1822 and later by Gruber [1], is caused by the failure of mesodermal induction. Of the 200 cases which have been reported so far, the highest incidence has been observed in the Gujarati Indians, with 1 affected birth per 1,300 (carrier rate 1 in 18) [2].
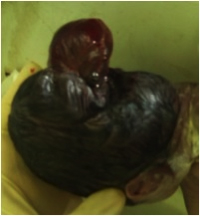
MGS is a lethal, rare and autosomal recessive condition that commonly occurs in association with chromosome 17 [2]. This syndrome is characterised by cystic kidneys, occiptal encephalocele and post axial polydactly. Two out of the three major anomalies are sufficient for the definitive diagnosis. Many congenital anomalies are present, of which the most striking is occipital encephalocele [3]. This cconsists of extrusion of parts of the brain parenchyma that is fully covered by a dural sac.
A 16–year–old primigravida presented in labour at 32 weeks of gestation with a preterm, premature rupture of membranes. A routine ultrasonography, which was done at 20 weeks, had diagnosed a foetus with several structural malformations that were suspected to be lethal. She continued the pregnancy on religious grounds. The ultrasonogram showed occipital encephalocele, polydactyly, oligohydramnios and cystic dysplasia of both kidneys. There was no history which was suggestive of an infectious origin for the malformations, but a genetic cause was suspected, as the patient had a consanguineous marriage. She delivered a 1.5 kg male foetus vaginally that had an occipital encephalocele [Table/Fig-1], a bell-shaped chest, a protuberant abdomen and post axial polydactyly of both hands and the left leg [Table/Fig-2]. On abdominal examination large masses were found, which were palpable in the lumbar regions. The baby died 2 hours after birth. A postmortem examination confirmed herniation of the brain and that both kidneys had multiple small cysts. A genetic analysis could not be done, as patient was unable to afford this investigation. The intra–natal and post–natal periods of the mother went on uneventfully. Based on the presence of classical features, a diagnosis of MGS was suggested.
Bell-shaped chest, protuberant abdomen and post axial polydactyly
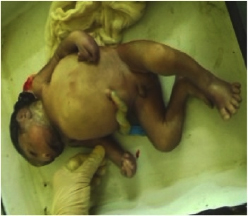
In MGS, the incidence of occipital encephalocele and post-axial polydactyly is 60% -80% and 55%-75%, respectively [2]. Renal disorders are noted in 95-100% of the cases. In this case, a foetal renal disorder like cystic dysplasia of bilateral kidneys was noted during autopsy; central nervous system defects and skeletal system malformations which included micrognathia were also present.
In MGS most of the infants with are stillborns or they die hours or days after birth, the implicated cause being dysplastic kidneys that result in oligohydramnios, which ultimately leads to foetal pulmonary hypoplasia [2]. The longest known survivor lived only till 4 months [4].
The possible differential diagnoses are trisomy13, autosomal dominant polycystic kidney disease(PKD) and autosomal recessive Smith-Lemli-Opitz (SLO) syndrome [5].
In view of the high recurrence rates of MGS, subsequent pregnancies should be properly investigated by first trimester ultrasound scans, with an early recourse to a chorionic villus biopsy or amniocentesis, if it is needed.
Though neonatal autopsy and genetic studies are gold standards for the diagnosis, detecting two out of the three major anomalies in the foetus, as has been mentioned above, is also sufficient for clinicians to arrive at a definitive diagnosis and for counselling the patients likewise in such hospital set-ups where neither is possible.
[1]. Jones KL, Smith’s Recognizable patterns of human malformation 1997 5th edPhiladelphiaW.B. Saunders:184-85. [Google Scholar]
[2]. Jayakar PB, Spiliopoulos M, Jayakar A, Meckel-Gruber Syndrome. eMedicine PaediatricGenetics and Metabolic Disease 2010 Jan [updated Sep 22, 2011]. Available from: http://emedicine.medscape.com/article/946672-overview [Google Scholar]
[3]. Baraitser M, Winter RM, Color Atlas of Congenital Malformation Syndromes 1996 1st edUKMosby-Wolfe:64 [Google Scholar]
[4]. Ramachandran U, Malla T, Joshi KS, Meckel-Gruber syndromeKathmandu University Medical Journal 2006 4(3(15)):334-36. [Google Scholar]
[5]. Visapaa I, Salonen R, Varilo T, Paavola P, Peltonen L, Assignment of the locus for hydrolethalus syndrome to a highly restricted region on 11q23-25Am J Hum Genet 1999 65:1086-95. [Google Scholar]