Bardet Biedel Syndrome: A Very Rare Entity in India
Rupal V. Dosi1, Nikita R. Bhatt2, Annirudh P. Ambaliya3, Nitin N. Sonune4, Rushad D. Patell5
1 Additional Professor, Medical College Baroda and Sayaji General Hospital, Vadodara, India.
2 Intern, Medical College Baroda and Sayaji General Hospital, Vadodara, India.
3 Assistant Professor, Medical College Baroda and Sayaji General Hospital, Vadodara, India.
4 Post graduate Student, Medical College Baroda and Sayaji General Hospital, Vadodara, India.
5 Senior Resident, Medical College Baroda and Sayaji General Hospital, Vadodara, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Rushad Patell, 32 Alka Society, Akota, Vadodara-390020, India Email: rushadpatell@gmail.com
Bardet Biedel Syndrome (BBS) is a rare autosomal recessive disease which is characterized by obesity, retinitis pigmentosa, polydactyly, neuro-developmental retardation and renal defects amongst others. It is a genetically heterogeneous ciliopathic disorder with inter and intra familial variations. Very few cases have been reported from India. We are reporting here a case of an adolescent girl who was diagnosed at the age of 16, with additional features of insulin resistance and non-alcoholic fatty liver disease. A review of recent literature and a short discussion on the care and management of this uncommon condition follow.
Bardet Biedel Syndrome, Retinitis pigmentosa, Obesity
Case Report
A 16–year–old girl presented to our department with complaints of progressive diminution of vision, excessive weight gain and learning disabilities. She was born of a consanguineous marriage and there was no history of similar complaints in the family. She was born at term by normal delivery, without any antenatal or post–natal complications. A learning disability was noticed in preschool and after a formal testing, she was found to have an IQ of 60. She was placed in the category of mild mental retardation. At the age of seven, she could no longer keep up and had to discontinue her schooling. Over the next 7–8 years, she gained excessive weight and developed dimness of vision, which had begun as a difficulty in seeing at night. Irregular menstrual cycles were also reported since she had attained menarche at the age of 13.
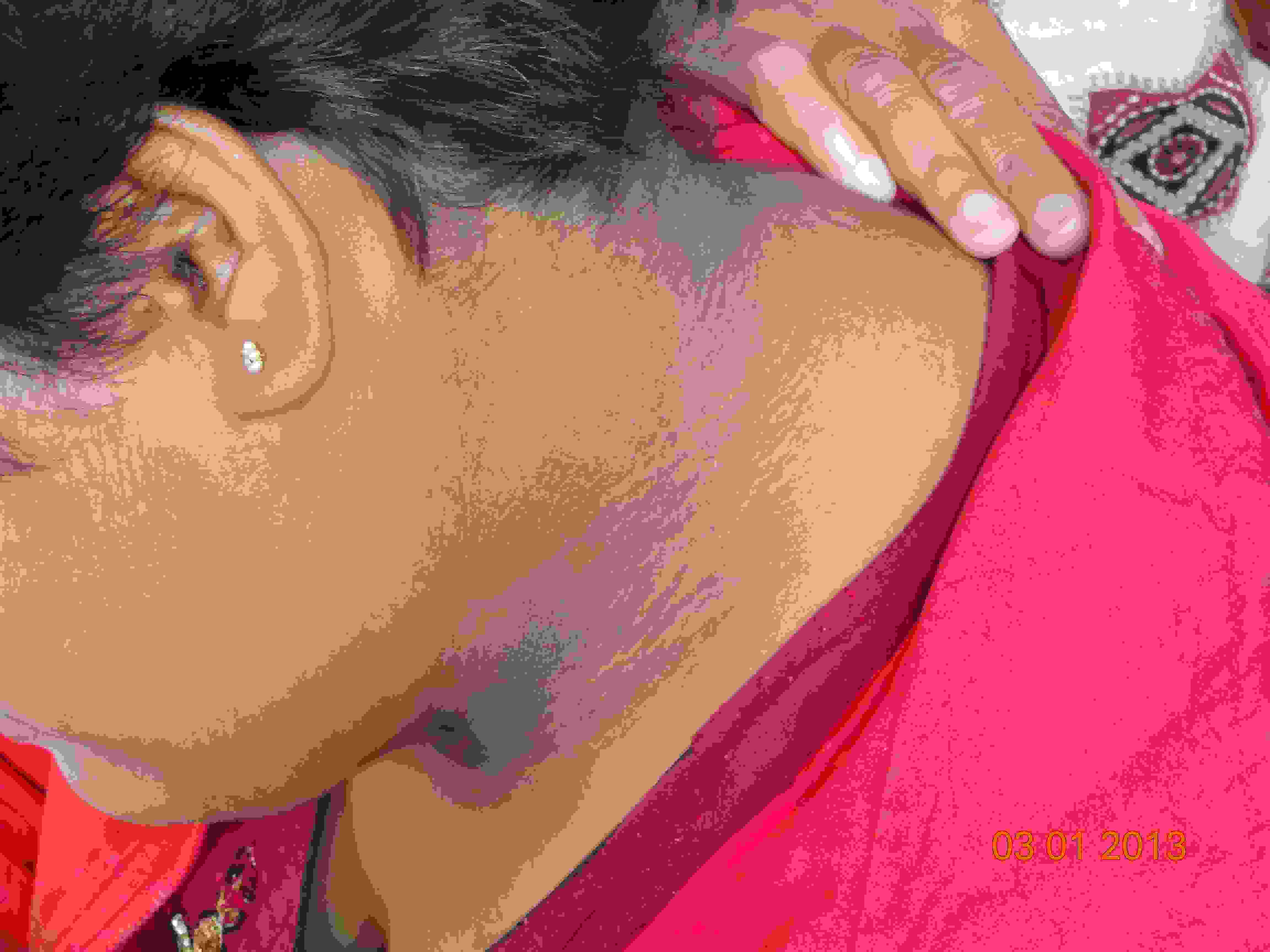
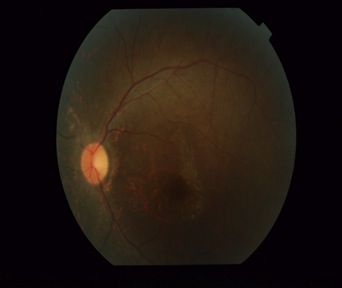
On examination, a marked truncal obesity was noted. Her height was 150 centimetres (below 3rd percentile according to the CDC stature for age chart) and her weight was 75kg (above 95th percentile according to the weight for age chart), with a BMI of 33.33 (above 97th percentile according to the BMI for age chart), which placed her in the obese category according to the WHO criteria. Moreover, she had a waist circumference of 105 cm (girls who are between 12 to 15 years of age have a mean normal circumference of 78.9 cm) and a hip circumference of 135 cm, resulting in a waist to hip ratio of 0.79, which best fitted the condition of central (abdominal) obesity. Her pulse rate was 90/min and her blood pressure was 150/92mm Hg. Postaxial polydactyly in the left upper limb [Table/Fig-1] and a velvety dark hyper-pigmentation in the neck folds, which were recognized as characteristic of acanthosis Nigricans, were also noted [Table/Fig-2]. Though her menstrual cycles were irregular, she had normal sexual and physical development and had attained Tanner stage four with normal breast development and pubic hair distribution and no hirsutism. Gynaecological examination was normal. Ophthalmic evaluation showed a visual acuity of 6/18 in left eye and of 6/36 in right eye. Fundus examination depicted disc pallor, attenuated retinal vessels and scattered pigment deposition, which were suggestive of retinitis pigmentosa [Table/Fig-3]. Rest of the systemic examination was unremarkable, apart from an obvious impaired intellect and garbled speech.
Extra–axial polydactyly noted in left upper limb

Black velvety hyperpigmentation noted in neck folds
Fundus showed, disc pallor, attenuated blood vessels and pigmentation, suggestive of retinitis pigmentosa
On undergoing laboratory investigations [Table/Fig-4], she was found to have a normal haemogram, her urine examination was normal and she had a normal renal profile. However, her aminotransferase levels were raised. Serum bilirubin and albumin levels and prothrombin time were normal. Her fasting glucose and oral glucose tolerance testing suggested prediabetes. Her lipid profile was normal. Her endocrinal work up was suggestive of subclinical hypothyroidism. However, prolactin levels and gonadotropins (FSH and LH) were appropriate for age and sexual development. Imaging studies depicted mild hepatomegaly with fatty changes on ultrasonogram of abdomen and normal kidneys and ovaries. Electrocardiogram and echocardiography were also normal.
Laboratory Investigations of Patient
| Laboratory test | Patients value | Reference range 9 |
|---|
| Hemoglobin | 11.1gm/ 100 ml | Females: 12-16g/100ml |
| Erythrocyte Sedimentation Rate | 32mm/hr | Females <50 yrs: 0-20mm/hr |
| Kidney Function Tests |
| Serum creatinine | 0.9mg/dl | 0.5-1.0 mg/dl |
| Blood urea nitrogen | 31mg/dl | 8-23mg/dl |
| Liver function tests |
| SGPT | 58mg/dl | 10-40mg/dl |
| SGOT | 66mg/dl | 10-40mg/dl |
| Total bilirubin | 0.8 mg/dl | 0.3-1.2mg/dl |
| Endocrine profile |
| Fasting glucose | 116mg/dl | 70-110mg/dl |
| PG2BS | 153mg/dl | <140mg/dl |
| TSH | 6.05 mIU/ml | 0.4-4.2 mIU/ml |
| Free T3 | 2.31pg/ml | 1.4-4.4 pg/mL |
| Free T4 | 0.97ng/dl | 0.9-2.3 ng/dL |
| FSH (Follicle Stimulating Hormone) | 3.92 mIU/ml | 1-100mIU/ml |
| LH (Lutenising Hormone) | 0.49 mIU/ml. | 1-104mIU/ml |
| Lipid profile |
| Total cholesterol | 120mg/dl | <200mg/dl |
| Triglyceride | 50 mg/dl | <130mg/dl |
| High density lipoprotein (HDL) | 38 mg/dl | 35-60mg/dl |
| Low Density Lipoprotein (LDL) | 72 mg/dl | 30-149mg/dl |
| Urine tests |
| Proteins in urine | Nil | Nil |
| Glucose in urine | Nil | Nil |
Based on the presentation, a familial cause of her obesity and mental retardation was thought of, and the additional findings of retinitis pigmentosa and polydactyly led to the recognition of Bardet Biedel Syndrome with insulin resistance and non-alcoholic fatty liver disease.
She was advised to wear glasses and to lose weight by exercising and by following a low calorie diet regimen. She was also given hormonal treatment for regulating her menstrual cycles. She was asked to come for regular follow-ups every 3 months, to monitor any further visual disturbances, to keep a watch on her menstrual cycles and to monitor the risk factors of metabolic syndrome, to prevent future development of diabetes and other cardiovascular problems.
Discussion
Bardet-Biedel syndrome was named after Georges Bardet and Arthur Biedel who noted a complete form of this syndrome for the first time. It is a rare ciliopathic pleiotropic autosomal-recessive disorder, with the frequency of its occurrence being approximately 1:1,60,000 [1]. Mutations in 14 genes are now known to be associated with BBS [2,3]. A greater incidence has been observed in communities with a high level of consanguinity, with a slightly higher prevalence in males (1.3:1) and the average age of diagnosis is 9 years [4].
The diagnosis of Bardet Biedel Syndrome is established on the basis of clinical criteria [Table/Fig-5] which have been suggested by Beales et al. The presence of four primary features or three primary features plus two secondary features is considered to be diagnostic [4]. Our patient also had additional features which were suggestive of hypertension, pre diabetes, acanthosis and NAFLD, which prompted a diagnosis of metabolic syndrome as per the ATP 2005 criteria. The presence of central obesity, impaired fasting glucose and cutaneous markers of marked acanthosis nigricans pointed to underlying insulin resistance. Moreover, it has been found that 45% of patients with BBS have Type 2 diabetes, thus underlying the need for regular screening and vigilance [4]. The multiple possible underlying mutations which are identified with this syndrome make genetic testing extremely laborious, expensive and of limited availability [5].
Diagnostic Criteria. Features in bold were identified in our patient
| Primary Features | Secondary Features |
|---|
| Retinal dystrophy | Speech delay/disorder |
| Post–axial polydactylyy | Developmental delay |
| Truncal obesity | Craniofacial dysmorphism |
| Hypogenitalism | CVS anomalies |
| Renal abnormalities | Hepatic involvement |
| Learning disabilities | Anosmia, ataxia, mild hypertonia |
BBS has an adverse prognosis, with an early onset of blindness, obesity, hypertension, and diabetes mellitus. Renal impairment is the major cause of morbidity/mortality in these patients, which goes undetected many times. The management of this syndrome is multi–disciplinary, which focuses on individual features. These patients should undergo surveillance in the form of regular ophthalmologic evaluations as well as blood pressure, lipid profile and diabetes mellitus testing, along with renal function monitoring on a regular basis [6].
A lack of medical awareness of this condition and the difficulty of making a firm diagnosis, owing to the slow emergence of features such as retinitis pigmentosa and renal dysfunction, contribute to the delay in the diagnosis and an early institution of therapy and surveillance [4]. This paper has highlighted the importance of having a high index of suspicion on genetic causes of childhood obesity and it aimed at increasing awareness in the medical community on this rare syndrome, especially in the Indian scenario.
[1]. Kumar S, Mahajan BB, Mittal J, Bardet-Biedl syndrome: A rare case report from North IndiaIndian J Dermatol Venereol Leprol 2012 78:228 [Google Scholar]
[2]. Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndromeNature 2003 425:628-33. [Google Scholar]
[3]. Adams M, Smith UM, Logan CV, Johnson CA, Recent advances in the molecular pathology, cell biology and genetics of ciliopathiesJ Med Genet 2008 45:257-67. [Google Scholar]
[4]. Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA, New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population surveyJ Med Genet 1999 36:437-46. [Google Scholar]
[5]. Adams M, Smith UM, Logan CV, Johnson CA, Recent advances in the molecular pathology, cell biology and genetics of ciliopathiesJ Med Genet 2008 45:257-267. [Google Scholar]
[6]. Sahu JK, Jain V, “Laurence-Moon-Bardet-Biedl Syndrome.”Journal of Nepal Medical Associatiom 2008 47.172:235-37.National Center for Biotechnology [Google Scholar]