Dentinogenesis Imperfecta : A Family which was Affected for Over Three Generations
Poornima Surendra1, Rohan Shah2, Roshan N.M.3, V.V. Subba Reddy4
1 Professor & HOD, Paediatric & Preventive Dentistry College of Dental Sciences, Davangere, Karnataka, India.
2 Post Graduate, Paediatric & Preventive Dentistry College of Dental Sciences, Davangere, Karnataka, India.
3 Reader, Paediatric & Preventive Dentistry College of Dental Sciences, Davangere, Karnataka, India.
4 Professor, Paediatric & Preventive Dentistry College of Dental Sciences, Davangere, Karnataka, India.
NAME, ADDRESS, E-MAIL ID OF THE CORESPONDING AUTHOR: Dr. Poornima Surendra, Professor & HOD, Paediatric & Preventive Dentistry,College Of Dental Sciences, Davangere, Karnataka, India.
Phone: (+91)8105482841,
E-mail: drpoornimas@yahoo.co.in
Dentinogenesis Imperfecta (DI) or hereditary opalescent dentin is inherited in a simple autosomal dominant mode with high penetrance and low mutation rates. It generally affects both the deciduous and the permanent dentitions. DI corresponds to a localized form of mesodermal dysplasia which is observed in the histo-differentiation. An early diagnosis and treatment are therefore fundamental, which aim at obtaining a favourable prognosis, since at late intervention makes the treatment more complex. We are presenting here a case of DI in which the disease affected the three generations of a family in India.
Dentinogenesis imperfecta, Osteogenesis imperfecta, Opalescent dentin, Deciduous dentition, Autosomal dominant
Introduction
Dentinogenesis Imperfecta is a genetic oral disease and it was probably first recognized by Barret in 1882 [1].The term, ‘dentinogenesis imperfecta’ was coined by Robert and Schour in 1939. DI has a reported incidence of 1:8000 whites in the United States. Dentinogenesis Imperfecta (DI) is a hereditary developmental defect of the dentin formation, which results in the appearance of opalescent teeth and which occurs in the absence of any systemic disorder.It has also been referred to as Capdepont’s teeth and hereditary opalescent dentin. It is inherited in a simple autosomal dominant mode, with high penetrance and a low mutation rate. It generally affects both the deciduous and the permanent dentitions. The classifications which are given by Witkop and by Shields, are well accepted, but they are not satisfactory. The genetic research on the DI type-1 has confirmed that osteogenesis imperfecta was a separate entity from DI and a single mutation of the DSPP gene is shown to cause DI type -2 and type-3. So, a modified classification was given by Neville [2]. An early diagnosis and treatment are therefore fundamental, which aim at obtaining a favourable prognosis, since a late intervention makes the treatment more complex. We are presenting a case of DI in which the disease affected three generations of a family in India.
Case Report
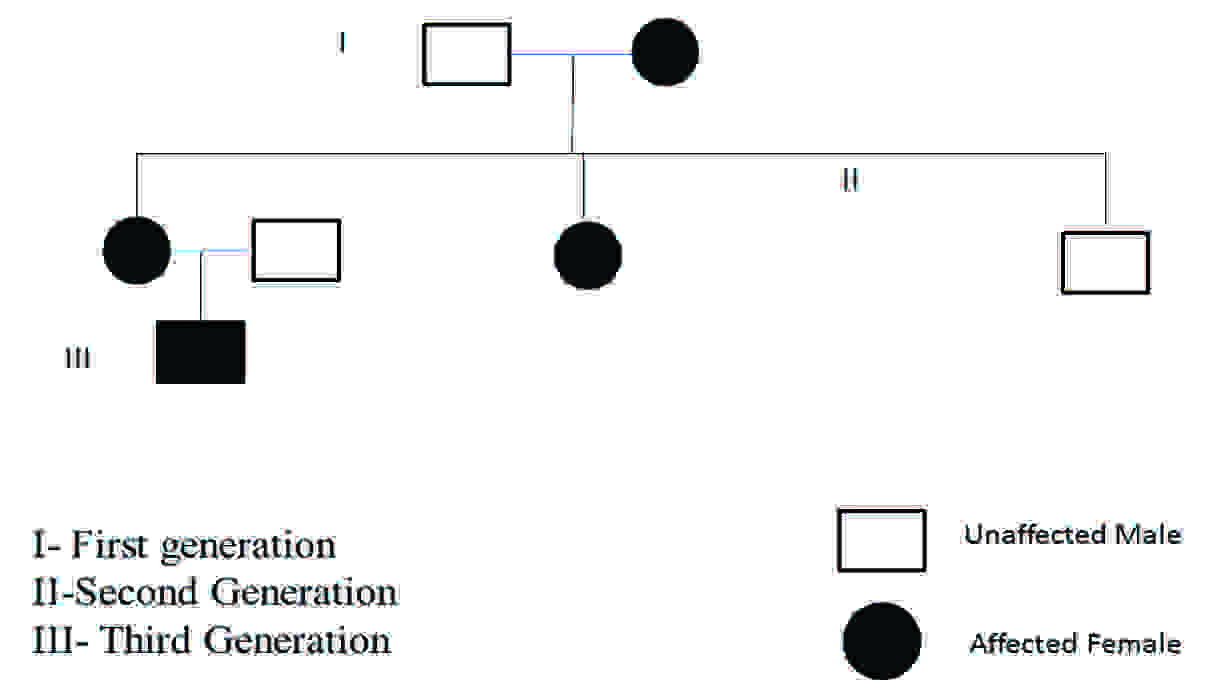
A 3–years old toddler visited the Department of Pedodontics and Preventive Dentistry, College of Dental Sciences, Davangere India, whose mother had the chief complaint of pain and unusually discoloured teeth. His history revealed gradually chipped teeth of the primary dentition. Due to a lack of diagnosis of the condition, no proper treatment was initiated. A family pedigree revealed that on his maternal side, his mother’s mother, mother’s sister, and his maternal grandmother were affected[Table/Fig-1]. His mother’s brother was not affected. The child’s mother and her sister, due to their lack of awareness on this disease, thus had received no timely treatment and they were wearing prosthesis. They were born to a non-consanguinous couple. No history of any type of bone abnormality was associated.
The Pedigree of Dentinogenesis Imperfect type II in the patient’s family:
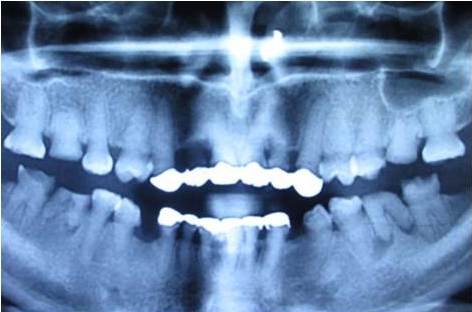
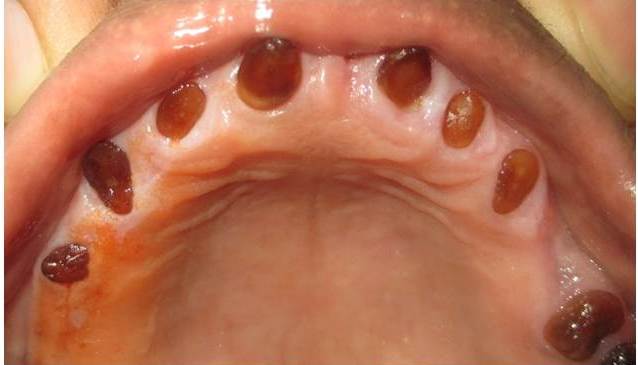
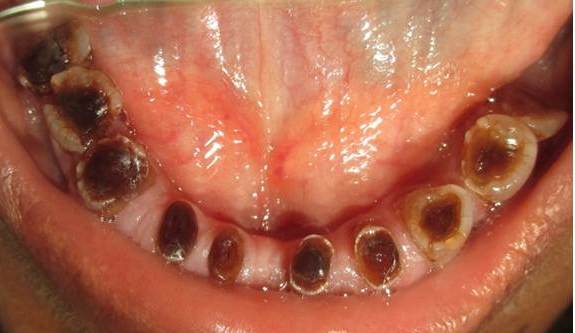
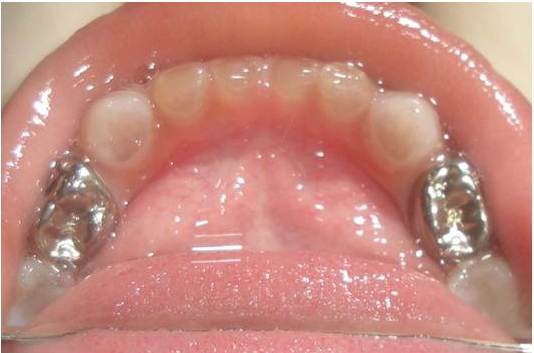
The clinical examination revealed loss of enamel and severe reduction in the vertical dimension. The teeth appeared brown, and most had worn incisal and occlusal surfaces upto the gingival level [Table/Fig-2]. The carious lesions which involved the pulp were associated with 74 and 84. The radiograph revealed severe attrition with no enamel, a wide canal with anterior teeth [Table/Fig-3] and a carious lesion approaching the pulp of 74 and 84, with incompletely formed roots [Table/Fig-4]. Panoromic radiographs were not taken, as the patient was very young and unco-operative. The patient’s mother’s panoromic radiograph and the IOPA revealed periapical radiolucencies which were associated with several teeth and short, blunted roots with marked cervical constrictions. The second molars had intact enamel and the pulp chambers were obliterated [Table/Fig-5,6,7 and 8]. The child’s maternal grandmother was also affected by the same condition [Table/Fig-9,10 and 11]. Based on the patient’s history, family history and the clinical and radiographic examinations, the conclusion of a DI variant ‘shell teeth’ in the child was made. MTA pulpotomy was performed with 74, 84 and on subsequent appointments stainless steel crowns were given [Table/Fig-12] [3,4]. The other treatments were difficult due to severe loss of the vertical dimension, the extreme fragility of the teeth and incompletely formed roots and as the patient was very young and un-coperative. The patient is under regular follow up.
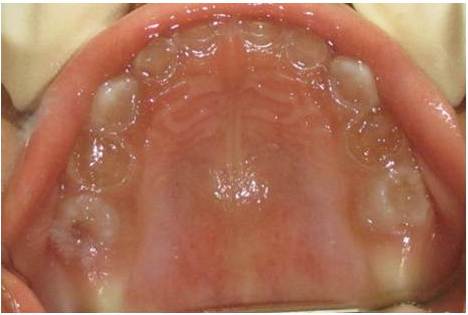
Intra-oral periapical radiograph of upper & lower anteriors
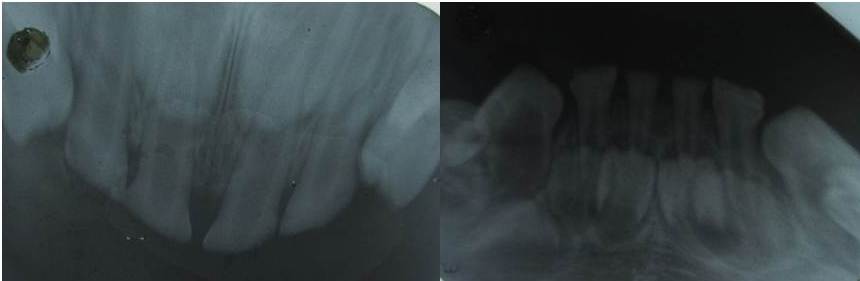
Intra-oral periapical radiograph of 74,75 & 84,85 region

Patient’s extra-oral view and Patient mother’s photo
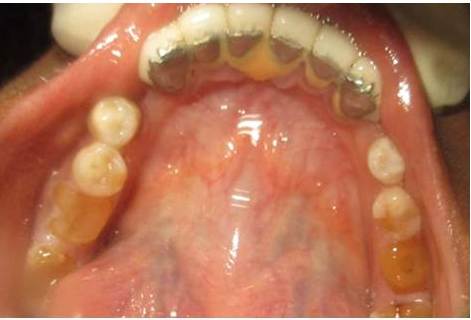
Patient mother’s upper arch with prosthesis
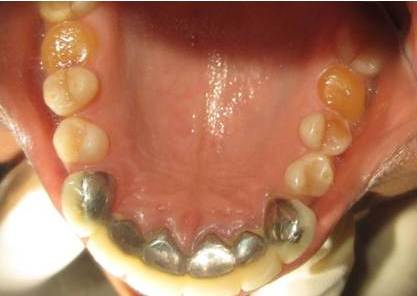
Patient mother’s lower arch with prosthesis
Patient grandmother’s photo
Patient grandmother’s upper arch
Patient grandmother’s lower arch
MTA pulpotomy followed by S.S crown
Differential Diagnosis
The differential diagnosis for Dentinogenesis imperfecta includes Dentin Dysplasia. Bulbous crowns, marked cervical constrictions, severe attritions, few periapical radioluscencies, and a premature teeth loss are the typical features of dentinogenesis imperfecta. So, based on the clinical and radiographic findings, a diagnosis of dentinogenesis imperfecta was made [5].
Discussion
The term, ‘Dentinogenesis imperfecta’ was coined by Robert and Schour in 1939. Earlier, Shields had divided dentinogenesis imperfecta into the DI type I, II and III defects. Witkop named the types as dentinogenesis imperfecta, hereditary opalescent dentin, and brandywine isolate. However, this classification has been changed after the recent genetic studies which have been done. It is a localized form of mesodermal dysplasia which is observed in the histo-differentiation [1,6,7] DI represents a basic defect in the structural or the regulatory proteins. The defective DI causing gene has been identified as Dentin Sialophosphoprotein (DSPP) and it has been mapped to chromosome 4q21.3. The gene product is a precursor protein that is cleaved into 2 dentin-specific proteins, Dentin Sialoprotein (DSP) and Dentin Phosphoprotein (DPP). The genetic basis for this clinical heterogeneity is unknown [8–12]. The extensive pedigrees of the individuals with DI, who were studied, had not exhibited changes which were suggestive of osteogenesis imperfecta. Osteogenesis imperfecta has been associated with a mutation of the COLL1AI/A2 gene, that encodes the production of type I collagen, whereas DI is associated with a mutation of the DSPP gene. Out of 8 mutations, 7 are associated with DI and the eighth is known to produce dentin dysplasia type II [2]. The examination of the family pedigree revealed that in each instance, an affected child had an affected parent. This indicated the complete penetrance of the gene in the family.
DI is characterized by opalescent teeth with marked attritions and short blunted roots which are constricted in the cervical regions [13]. The discolouration in the primary dentition can be classified as yellow/brown or opalescent gray. The yellow/brown DI is more prevalent and more prone to attrition than the opalescent gray DI [14]. This disorder may involve either the primary or the permanent dentitions or both, with the association of the shell teeth, but it is more common in the primary teeth. The severity of the trait in the individual teeth, varies with the age at which the particular tooth develops. The histologically affected teeth demonstrate altered dentin, with the atypical granular dentin matrix demonstrating an interglobular calcification. The enamel is normal in most of the patients; however, one third of the patients have hypoplastic or hypocalcified defects [2].
The essential aim of treating this disorder is to prevent the attrition of the erupted tooth and to establish proper vertical dimensions [5]. This will enable an early diagnosis and treatment, so as to intercept the attrition and to preserve the function, aesthetics and the normal growth. It has also been said that in those with DI in the primary dentition, DI may be absent in the permanent dentition. But interestingly, those with DI in the permanent dentition always were found to have DI in the primary dentition [14]. Severe DI can be treated at two stages in the deciduous teeth. If it is diagnosed early, stage I is at around 18-20 months and stage II can be at around 28-30 months [14]. This will also help the disease to be curbed at its onset in the deciduous dentition and it can pave the way for a healthier permanent dentition. It is also necessary for the affected ones to go for a genetic councelling.
Conclusion
The patients with the DI condition present with different degrees of severity. A comprehensive interdisciplinary treatment planning is required to rehabilitate these patients. This present case have been reported as an attempt on our part to create awareness among the general dentists, so that they can make the common masses aware of these genetic oral diseases.
[1]. Raji MA, Varghese NO, Grorge K, Dentinogenesis imperfecta. Report of three cases in an Indian familyIndian J Dent Res 1993 4:59-64. [Google Scholar]
[2]. Abnormalities of the Teeth, Neville, Textbook of oral and maxillofacial pathology, third edition, chapter II pg no 106-09 [Google Scholar]
[3]. Kindelan SA, Day P, Nichol R, Willmott N, Fayle SA, British Society of Paediatric Dentistry. UK National Clinical Guidelines in Paediatric Dentistry: stainless steel preformed crowns for primary molarsInt J Paediatr Dent 2008 18(Suppl 1):20-28. [Google Scholar]
[4]. Kilpatrick NM, Welbury RR, Advanced restorative dentistryIn: Paediatric Dentistry 2005 OxfordOxford University Press:213Welbury RR, Duggal MS, Hosey MT, eds [Google Scholar]
[5]. Barron Martin J, McDonnell Sinead T, MacKie Iain, Dixon Michael J, Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasiaOrphanet Journal of Rare Diseases 2008 3:31 [Google Scholar]
[6]. Modesto A, Alves AC, Vieira AR, Portella W, Dentinogenesis imperfecta type II: case reportBraz Dent J 1996 :7.47-52. [Google Scholar]
[7]. Huth KCH, Paschos E, Sagner T, Hickel R, Diagnostic features and pedodontic-orthodontic management in dentinogenesis imperfecta type II: a case reportInt J Pediatr Dent 2002 12:316-21. [Google Scholar]
[8]. Lee S K, Lee K E, A Novel Mutation in the DSPP Gene Associated with Dentinogenesis Imperfecta Type IIJ Dent Res January 2009 88:51-55. [Google Scholar]
[9]. Haihua Bai1, Hasi Agula, A novel DSPP mutation causes dentinogenesis imperfecta type II in a large Mongolian familyBMC Medical Genetics 2010 11:23 [Google Scholar]
[10]. Malmgren B, Lindskog S, Elgadi A, Norgren S, Clinical, histopathologic, and genetic investigation in two large families with dentinogenesis imperfecta type IIHum Genet 2004 14:491-98. [Google Scholar]
[11]. Kim JW, Nam SH, Jang KT, Lee SH, Kim CC, A novel splice acceptor mutation in the DSPP gene causing dentinogenesis imperfecta type IIHum Genet 2004 115:248-54. [Google Scholar]
[12]. Holappa H, Nieminen P, Tolva L, Lukinmaa P L, Alaluusua S, Splicing site mutations in dentin sialophosphoprotein causing dentinogenesis imperfecta type IIEur J Oral Sci 2006 114:381-84. [Google Scholar]
[13]. Kantaputra PN, Dentinogenesis imperfecta-associated syndromesAm J Med Genet 2001 104:75-78. [Google Scholar]
[14]. Sapir S, Shapira J, Dentinogenesis imperfecta: an early treatment strategyPediatr Dent 2001 23:232-37. [Google Scholar]