Holoprosencephaly (HPE), a disorder which results from a failure of cleavage or the incomplete differentiation of the forebrain structures at various levels or to various degrees, is related to hereditary factors, chromosomal anomalies, cytogenetic abnormalities, and environmental teratogenic factors. We are reporting a case of a multiparous woman who was G3,P3,L2, who delivered a full term foetus with holoprosencephaly and multiple craniofacial anomalies. An autopsy was conducted. Multiple anomalies of the craniofacial bones, which include hypoplasia and synostosis of the frontal bone, anophthalmia, absence of the anterior cranial fossa, hypoplasia of the maxillae, an absent antrum, cleft palate, a central hare lip and arrhinia which includes absence of the nostrils and hypotelorism of the eye placodes, were noted. This case is being reported for its rarity and the available literature was reviewed in this respect.
Case Report
A 24 years old multiparous lady who was G3, P3, L2, attended the antenatal outpatient department at 28 weeks of gestation. A screening antenatal ultrasonography was done and it was found that baby had HPE. She was advised termination of the pregnancy, but she refused and continued with the pregnancy. At term, she presented with foetal distress. She was admitted to the hospital and a female baby which weighed 2500 grams was delivered by craniotomy. After the craniotomy, the foetus survived for few hours and then died. The dead foetus was sent to the pathology laboratory for an autopsy study with the consent of the mother. There was no history of a consanguineous marriage. The first pregnancy was a full term, normal vaginal delivery and it was a female child. She had attained normal mile stones but had succumbed to viral fever at 1 year of age. The second child was a male, who was delivered by a normal vaginal delivery, had attained normal mile stones and was alive. The third pregnancy was the present case, and the patient had conceived 2 years after she had delivered the second child. The patient had no history of any medical illness which included diabetes or hypertension and she had not used any teratogenic drugs.
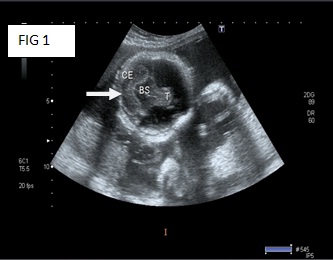
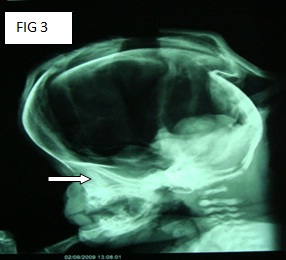
An ultrasound of the pelvis of the mother, which was provided along with the body of the baby, showed the following results: An ultrasound of the pelvis was done at 28 weeks of gestation. It showed a foetus with a monoventricle, with no obvious neural tissue in the region of both the cerebral hemispheres. The falx cerebri and the inter hemispheric fissures could not be visualized. Both the thalami were fused. The cerebellar hemispheres could be seen [Table/Fig-1]. The nasal bridge was depressed. A cleft lip was seen. The impression was that of a single live foetus in a cephalic presentation, at 28 weeks and 2 days of gestational age, with alobar holoprosencephaly.
Ultra sound of foetus at 28 week of gestation monoventricle with no obvious neural tissue
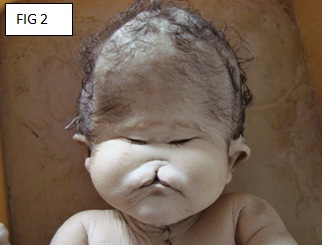
Autopsy: A dead female foetus, along with the placenta, was received. The weight of the foetus was 2.5 kgs. The total length was 45 cms, the crown-rump length was 30 cms, the head circumference was 12.5 cms, the chest circumference was 13 cms, and the abdominal circumference was 12.5 cms. The external examination of foetus showed an enlarged head, arrhinia, absence of the nostrils, hypotelorism of the eye placodes and absence of the cornea or the sclera. Dissection of the soft tissue which was underneath the eye placodes showed the absence of both the eye balls. The examination of the oral cavity showed a cleft palate and a central hare lip [Table/Fig-2 & 3].
Arrhinia, absence of nostrils, hypotelorism of eyes, central hair lip
X-ray of skull -Hypoplasia of Maxillae & absence of Maxillary Sinuses
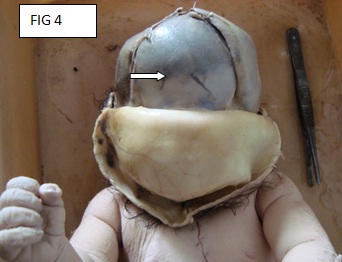
Single hypoplastic Frontal Bone with absence of metopic sutures
Anopthalmia. Eye balls replaced by rudimentary softissue mass
A midline incision was made from the symphysis menti to the symphysis pubis, and the body was opened. All the body cavities were examined. No situs inversus was seen. The organs were removed enblock. All the internal organs were grossly normal for the gestational age, except the adrenals. One adrenal was hypoplastic (3grams) and the other adrenal was absent.
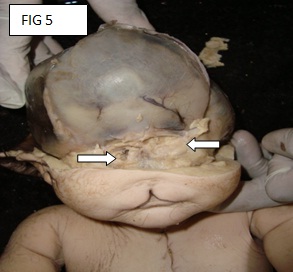
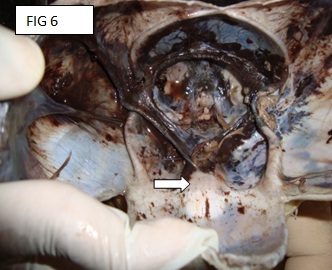
There is a rent in the scalp, which was suggestive of an anterior craniotomy. The skull was opened by making a coronal incision of the scalp which joined both the mastoid processes. The calvarium was opened through the fibrous joints of the parietal, frontal and the occipital bones. The anterior fontenellae was already open. The brain was not seen (the foetus was delivered by craniotomy). The examination of the skull showed the absence of the anterior cranial fossa and a single hypoplastic frontal bone with the absence of the metopic sutures [Table/Fig-4 & 5]. No optic chiasma or olfactory bulb was seen. The pituitary fossa was not seen.
Absent anterior cranial fossa
The examination of the rest of the bones, which was done by X-ray, showed hypoplasia of the maxillae, absence of both the maxillary antrum on both the sides and absence of the nasal bridge [Table/Fig-6]. The microscopic examinations of both the lungs showed aspiration of the amniotic fluid in the focal areas, which in some of the alveoli, was seen as squames, refractile lipid bodies and lanugo hair. One adrenal was smaller in size and it showed the focal nodular patterns of the fasciculate and the reticularis cells. The other adrenal was not present. The microscopic study of rest of the organs was normal.
The placenta weighed 525 grams, its size was 17x16.5cms, it was fully developed, and it had 12 cotyledons. The chorioamniotic membranes were normal and thin. The length of the umbilical cord was 35 cms, it was eccentrically inserted and the number of vessels were 3. The placental histology did not show any abnormality.
Based on the antenatal ultrasound findings, the radiological findings, the gross appearances and the microscopic appearances, the diagnosis of a full term, mature female child, with multiple external congenital anomalies, adrenal hypoplasia and holoprosencephaly was made.
Discussion
Holoprosencephaly is a disorder which is caused by the absence or the incomplete diverticulation and cleavage of the embryonic forebrain (prosencephalon) into the cerebral hemispheres and the lateral ventricles. This leads to defects in the development of the brain structure and function and also in the development of the face. In 1963, Demyer et al., [1] proposed the term, ‘Holoprosencephaly’(HPE). Normally, the forebrain is formed and simultaneously, the face begins to develop in the fifth and the sixth weeks of the human pregnancy [2]. In the case which has been presented above, the gestational age during an ultrasound study was 28 weeks, thus confirming HPE. The Hedgehog signaling pathway plays a critical role in the morphogenesis of these structures, and the loss-of-function or mutations of the individual components within this pathway are reported in families with a history of recurrent HPE. The mutations which affect the sonic hedgehog or its signaling pathway may result in HPE [3] The incidence of HPE is 1:250 during embryogenesis and it is 1:16000 of all the live births [4]. HPE is an aetiologically heterogeneous disorder and about 30-40% of all the affected individuals have an underlying chromosomal disorder [5]. It is usually associated with an abnormality of chromosome 13 (trisomy 13, deletion or duplication of 13q or ring 13). The other associated chromosomal abnormalities include trisomy, deletion 2p, duplication 3p, deletion 7q, etc. HPE can also occur as a component of multiple malformation syndromes like the Meckel’s syndrome, the Oral-facial-digital syndrome type VI, the Pallister-Hall the Syndrome and Smith-Lemli-Opitz syndrome [2, 6]. About 1-2% of the infants who are born to diabetic mothers have HPE [7]. In our case, the mother was non diabetic. Isolated cases with a Mendelian inheritance are also seen. These are usually transmitted as an autosomal dominant trait with a variable penetrance and a wide familial variance [3]. In these cases, the parents may have only mild cranio-facial abnormalities like a single central maxillary incisor. For the autosomal dominant, non-syndromal HPE, four genes have been identified so far. The disorder is found twice as often in female babies [8]. In our case also, the foetus was a female.
HPE can be classified, depending on the degree of abnormality [2]. In alobar HPE, which is the most severe of the three types, a single ventricular cavity is present, the thalami are fused and the corpus callosum, the falx, the inter-hemispheric fissure, the optic tracts and the olfactory bulbs are absent. In our case, the ultrasound showed a monoventricle with no obvious neural tissue, thus correlating our findings with the findings of other studies. The associated mid-facial defects in alobar HPE include cyclopia, ethmocephaly, cebocephaly and cleft lip/palate, which are associated with hypotelorism. In our case, the foetus showed an enlarged head, arrhinia, absence of the nostrils, hypotelorism and absence of the eyes. In semi-lobar HPE, the occipital horns of the ventricles are partially formed and the posterior portions of the falx and the inter-hemispheric fissures are seen. The thalami may only be partially fused. Anteriorly, the ventricles are fused and the corpus callosum, the optic tracts and the olfactory bulbs are absent. The facial defects are milder and cyclopia and ethmocephaly are not usually seen. In the mildest form-lobar HPE, the fusion is partial, with the presence of an anterior cleavage. The corpus callosum, the inter-hemispheric fissure and the falx are not fully developed and the roof of the anterior end of the frontal horn looks squared [3]. Our present case corresponded to the alobar form of HPE.
When the embryo’s forebrain does not divide to form the bilateral cerebral hemispheres (the left and right halves of the brain), it causes defects in the development of the face and in the brain structure and function [2, 4]. In our case also, multiple craniofacial anomalies were seen, as have been described above.
In a classic article, DeMyer et al., [1] discussed a graded series of facial anomalies that occur along with holoprosencephaly [Table/Fig-7]. The face predicts the development of the brain approximately 80% of the times. In the other 20% of the times, the facial features are non-diagnostic [9,10]. Barr and Cohen [11] reported essentially normal faces in some autosomal recessive cases of HPE. In the present case, the facial anomalies correlated with the features of HPE and hence, they were predictive of the brain development.
| Facial typeb | Main facial features | Brain |
|---|
| Cyclopia | Median monophthalmia, synophthalmia, or anophthalmia. Proboscis may be single or absent. Hypognathism in some cases. | Alobar holoprosencephaly |
| Ethmocephaly | Ocular hypotelorism with proboscis | Alobar holoprosencephaly |
| Cebocephaly | Ocular hypotelorism and blind-ended, single- nostril nose. | Usually alobar holoprosencephaly |
| Median cleft | Ocular hypotelorism, flat nose, and median cleft lip. | Usually alobar holoprosencephaly |
| Less severe facial dysmorphism | Variable features including ocular hypotelorism or hypertelorism, flat nose, unilateral orbilateral cleft lip, iris coloboma, or Other anomalies. Minimal facial dysmorphism insome cases. | Semilobar or lobar holoprosencephaly |
In the less severe cases of prosencephaly, the babies are born with a normal or near-normal brain development and facial deformities that may affect the eyes, nose, and the upper lip are seen. In our case, the baby was born alive, with facial anomalies. The extent of the brain development could not be assessed at the time of birth, since the mother did not report for antenatal checkups after 28 weeks and was admitted to the hospital in labour. Hence, the ultrasound which had to be done at 36 weeks was not available. In view of the previous ultrasound reports, the baby was delivered by craniotomy.
Cyclopia, one of the most severe type of the facial defects, is an abnormality which is characterized by the development of a single eye, which is located in the area which is normally occupied by the root of the nose, and a missing nose or a nose which is in the form of a proboscis (a tubular appendage), which is located above the eye. This condition is also referred to as cyclocephaly or synophthalmia, and it is very rare. Our case did not show cyclopia,; instead it showed hypotelorism and absence of the eye balls. No nasal bridge or an external nose was seen in contrast to the proboscis.
The most severe cases of HPE end up as miscarriages or stillbirths. Our case, however, is an exception to the above facts. Though it was alobar HPE with severe facial anomalies, the baby survived till full-term and after birth also, it survived for 3 hours and 45 minutes.
Conclusion
A case of holoprosencephaly with multiple craniofacial anomalies has been presented for its rarity. Certain interesting features were noted in this case; namely, the baby survived till term and the baby was live born even after the craniotomy. The HPE was of the alobar type and the craniofacial anomalies were of the severe type. In addition, the development of the eyes and nose were absent. Ultrasonography (USG) helps in detecting these anomalies at an early stage. USG and autopsy, together strengthen the knowledge in their respective fields. The documentation of such rare types of anomalies helps in further detailed studies on anomalies.
[1]. De Myer W E, Zeman W, Palmer C G, The face predicts the brain: diagnostic significance of median facial anomalies for holoprosencephaly (arhinencephaly)Pediatrics 1964 3:256-63. [Google Scholar]
[2]. Ashwal S, Congenital structural defects of the brain. In: Levene MI, Chervenak FA, Whittle M, edsFetal and Neonatal Neurology and Neurosurgery 2001 3rd ednLondonChurchill Livingstone:210-50. [Google Scholar]
[3]. Thomas NA, Cherian A, Sridhar S, HoloprosencephalyImages In Medicine 2003 2(49):173-74. [Google Scholar]
[4]. Cohen MM Jr, Perspectives on holoprosencephaly: Part I. Epidemiology, genetics, and syndromologyTeratology 1989 40:211-35. [Google Scholar]
[5]. Olsen CL, Hughes JP, Youngblood LG, Sharpe-Stimac M, Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989Am J Med Genet 1997 73:217-26. [Google Scholar]
[6]. Croen LA, Croen Lisa A, Shaw Gary M, Lammer Edward J, “Risk Factors For Cytogenetically Normal Holoprosencephaly in California: A Population-Based Case-Control Study”American Journal of Medical Genetics (Wiley-Liss) 2000 90(4):320-25. [Google Scholar]
[7]. Barr M Jr, Hanson JW, Currey K, Sharp S, Toriello H, Schmickel RD, Holoprosencephaly in infants of diabetic mothersJ Pediatr 1983 102:565-68. [Google Scholar]
[8]. Mollerlokken G, Brains and faces in holoprosencephaly: pre- and postnatal description of 30 casesUltrasound Obstet Gynecol 2002 19:24-38. [Google Scholar]
[9]. De Myer WE, Holoprosencephaly (cyclopia-arhinencephaly). In: Vinken P J, Bruyn G W, editorsHandbook of Clinical Neurology 1977 AmsterdamNorth-Holland Publishing Co:431-78. [Google Scholar]
[10]. Cohen MM Jr, Perspectives on holoprosencephaly. Part III. Spectra, distinctions, continuities, and discontinuitiesAm J Med Genet 1989 34:271-88. [Google Scholar]
[11]. Barr M Jr, Cohen MM Jr, Autosomal recessive alobar holoprosencephaly with essentially normal facesAm J Med Genet 2002 112:28-30. [Google Scholar]