The Prune Belly Syndrome in a Female Foetus with Urorectal Septum Malformation Sequence: A Case Report on a Rare Entity with an Unusual Association
Dibyajyoti Goswami1, Giriraj Kusre2, Hemonta Kumar Dutta3, Adity Sarma4
1 Post Graduate Trainee, Department of Anatomy, Assam Medical College and Hospital, Dibrugarh, Assam-786002, India.
2 Associate Professor, Department of Anatomy, Assam Medical College and Hospital, Dibrugarh, Assam-786002, India.
3 Associate Professor, Department of Paediatric Surgery, Assam Medical College and Hospital, Dibrugarh, Assam-786002, India.
4 Associate Professor, Department of Pathology, Assam Medical College and Hospital, Dibrugarh, Assam-786002, India.
NAME, ADDRESS, E-MAIL ID OF THE CORESPONDING AUTHOR: Dr. Giriraj Kusre, Assocoate Professor, Department of Anatomy, Assam Medical College and Hospital, Borbari, Dibrugarh, Assam-786002, India.
E-mail: giriraj.kusre@rediffmail.com
The prune belly syndrome is a rare congenital anomaly which is characterized by the triad of an absent or a deficient development of the abdominal muscle, bilateral cryptorchidism and an anomalous urinary tract. In its full form, this condition occurs only in males. However, a similar condition occurs in females in the absence of cryptorchidism. On the other hand, the urorectal septum malformation sequence is a lethal congenital malformation which is characterized by the development of a phallus like structure, a smooth perineum and the absence of urethral, vaginal and anal openings. We are reporting a case of a female foetus with the prune belly syndrome, which was associated with a urorectal septum malformation sequence. A dead foetus with a protruded abdomen and ambiguous genitalia, was born at 32 weeks of pregnancy. On autopsy, it was found to have female internal genital organs. The left kidney, the urinary bladder and the rectum were absent. The sigmoid colon, the ureters and the fallopian tubes opened into a common cloacal sac. The histopathological examination of the ovary showed the presence of Leydig’s cells. The occurrence of the female counterpart of the prune belly syndrome is extremely rare and only few of such cases were found to be discussed in the details in the indexed English literature so far. Hence, we hope that this case report will contribute to the existing knowledge on the prune belly syndrome.
Abdominal wall, Autopsy, Cryptorchidism, Prune belly syndrome, Leydig’s cells
Introduction
The Prune Belly Syndrome (PBS) or the Eagle – Barrett Syndrome is a rare congenital abnormality of unknown aetiology, with the characteristic triad that includes an absent or a deficient development of the abdominal muscle, which causes the skin of the abdomen to wrinkle like a prune, bilateral cryptorchidism and an anomalous urinary tract [1]. It was first recognized by Frohlich in 1839 and the name PBS was coined by Osler in 1901 [2]. The incidence of this condition has been estimated to be 1 in 29,000 to 50000 neonates [3]. By definition, the PBS can occur in its full form only in boys. Yet, very rarely, a similar condition can be present in girls in the absence of undescended testes [4]. The Urorectal Septum Malformation Sequence (URSM) or the cloacal dysgenesis sequence is a lethal congenital malformation which is characterized by the development of a phallus like structure, a smooth perineum and the absence of urethral, vaginal and anal openings as the primary malformations [5]. There persists a common cavity for the urinary, gastrointestinal and the genital structures [6]. The incidence of URSM is one in 50000 to 250000 neonates [7].
The PBS often coexists with a spectrum of extra–renal malformations. These include cardiac, gastrointestinal, pulmonary and skeletal anomalies. The gastrointestinal anomalies were reported to be associated with as high as 30% cases of the PBS [8]. Herein, we are reporting a case of the PBS in a female foetus, which was associated with the URSM sequence.
Case Report
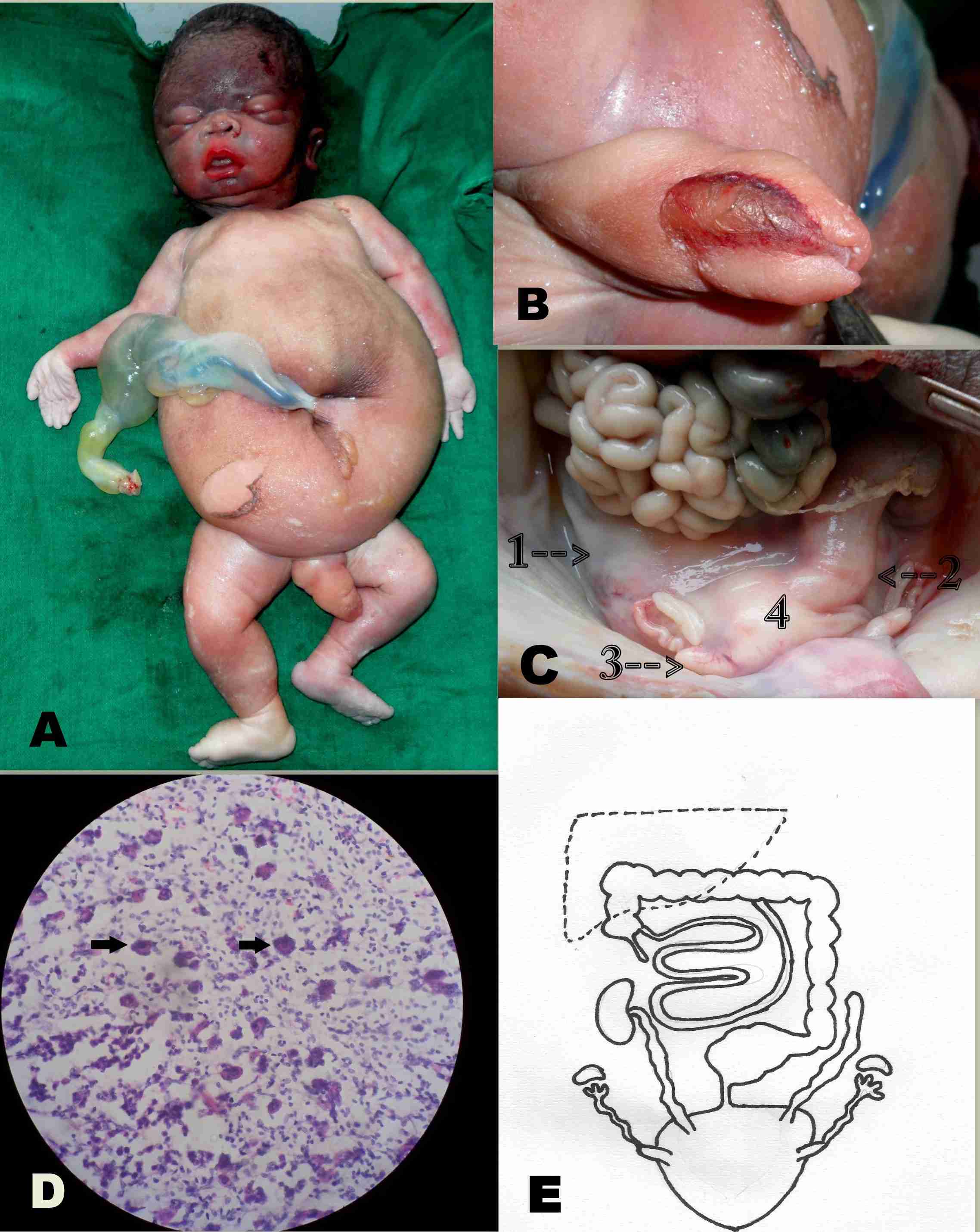
A 22-years old, healthy female from the north east region of India, who was gravida 2 and parity 1, who had a past history of epilepsy in her previous pregnancy, gave birth to a still born infant with ambiguous genitalia at 32 weeks of gestation. The previous child was a healthy male. There was no history of consanguity in the family or of an exposure to any known teratogen. The baby weighed 2.8kg. On external examination, the face of the infant was found to be rounded and the abdomen appeared flabby [Table/Fig-1A]. The perineum was smooth, without any anal or vaginal opening. The baby had a small phallus that resembled an enlarged clitoris, which was covered with a visible mucus membrane on its dorsal aspect [Table/Fig-1B]. The protruded skin on both sides of the mucus membrane gave an impression of labial folds. The other external features which included the eyes, ears, nose, lips, palate, fingers, toes and the vertebral column were normal.
Prune Belly Syndrome. External appearance of the fetus showing protruded abdomen (A) and Ambiguous genitalia (B). Ureter, fallopian tubes and dilated sigmoid colon (1,2,3 of C). Note that all these structures opened into the common cloacal sac (4 of C). Histological slide of ovary under microscope (D) showed Leydig cells indicated by arrow marks. Diagrammatic representation of autopsy findings (E) showing subhepatic caecum and absence of left kidney and dilated ureters. Sigmoid colon, fallopian tubes and ureters communicating to a common cloacal sac.
Autopsy Findings
On autopsy, the abdominal wall was found to be deficient of any muscular layer. The caecum was subhepatic in position. An abnormally dilated sigmoid colon was filled with calcified meconium that appeared as a yellowish putty like material. It opened with a constriction into a large pouch like structure (the common cloacal sac) [Table/Fig-1C]. The sac had two projections at the antero superior aspect, which were identified as the cornu of the uterus. At the other end, each cornu was connected to the fallopian tubes. The ovaries were present at their normal positions. There was no rectum and urinary bladder. The left kidney was absent and the upper end of the left ureter ended blindly. The right kidney was at its normal position. Both the ureters were dilated and tortuous and they drained into the common sac. One end of the sac was attached to the perineum, without any external opening.
Histological Examination
Tissues from the suspected cornu of uterus, the fallopian tubes, ovary, sigmoid colon (constricted portion), kidney and the ureters were subjected to histopathological examinations. Histology confirmed the identity of the respective organs, except that the ovary had stromal tissue with vascular elements and groups of cells with eosinophilic cytoplasm, central nuclei, and prominent nucleoli, which were identified as Leydig’s cells [Table/Fig-1D]. Moreover, the tissues from the sigmoid colon, near the constriction, were devoid of the ganglion cells. A cytogenetic examination could not be done due to the non availability of suitable samples.
Discussion
The present case had two out of the three characteristic features of the PBS viz. abdominal wall muscle deficiency and urinary tract abnormalities in the form of dilated ureters and an absent left kidney. It also had ambiguous genitalia with female internal genital organs and the third feature of the PBS i.e. cryptorchidism was absent. The female internal genital organs consisted of a bicornuate uterus, fallopian tubes and ovaries, which were histologically confirmed. So, the present case was diagnosed as a female counterpart of the PBS. The female counterpart of the PBS has been reported from time to time, though its incidence is quite low and accounts for 3-5% of all the recorded cases [9]. The embryogenesis of the PBS was not known precisely. However, three theories have been put forward to explain the various anomalies which are associated with this syndrome. The first theory explains an early uterourethral obstruction as the cause of the urinary tract dilatation and the foetal ascites. Next is the mesodermal arrest theory. The third theory mentions that a defect in the yolk sac is a possible cause [10]. In our case, the histological examination of the ureters showed normal tissue. Their dilatations were probably secondary to the obstruction which was distal to the common pouch. In the PBS, the primary cause of the megaureters was believed to be a urethral obstruction that subsequently results in dilatations of the ureters and ascites, leading to degeneration of the abdominal wall musculature [11].
An exceptional finding which was observed was the presence of Leydig’s cells in the histological sections of the ovary. It has been reported that an ectopic appearance of the Leydig’s cells in the ovaries can lead to masculinization of the female foetus due to the increased androgen secretion [12]. The phallus like structure which was seen in the present case was the enlarged clitoris, which had probably resulted from the effect of the androgen which was secreted by the Leydig’s cells of the foetal ovaries.
The sigmoid colon was filled with calcified meconium. The presence of calcified meconium in foetal bowel was an evidence of mixing of the meconium with urine due to a communication between the gastrointestinal and the urinary systems [13]. Escobar et al., first described the specific pattern of the developmental anomalies of the urogenital tracts and the lower intestinal tract, which were associated with a lack of the perineal opening and the presence of ambiguous genitalia as the URSM sequence [7]. In our case, all these features were present. The present case also had a dilated sigmoid colon with a distal constricted portion. The histology of the constricted portion showed the absence of the ganglion cells. In addition to this, the part which was distal to the constriction, terminated into a common cloacal sac that formed the common outlet for both the ureters and the two horns of the uterus. The sac in turn, was connected to the perineum, without any external opening. We are aware of our limitations. We did not perform a cytogenetic study for the sex determination. However, the presence of the female internal genital organs was sufficient to determine the sex of the foetus.
Our findings brought us to the conclusion that the foetus was born as a female equivalent of the prune belly syndrome, which was associated with a urorectal septum malformation sequence. The occurence of the PBS in females is rare. Only five female cases of the PBS were reported in a study which was done on half a million live births in British Columbia [14]. Moreover, its association with the urorectal septum malformation sequence makes it an extremely rare entity. Only a few cases of the PBS in female foetuses have been discussed in detail so far. A chance for a meticulous autopsy evaluation should never be missed in stillborn babies with obvious congenital anomalies. This is worthwhile for the confirmation and the revelation of other associated anomalies, as well as to help in understanding the embryogenesis of their formations. This case has been reported with the hope of contributing to the existing knowledge which pertains to PBS.
[1]. Hendren WH, Carr MC, Adams MC, Megaureter and Prune belly syndrome. In: O’Neill JA, Rowe MI et al editorsPaediatric surgery 1998 5th editionMosby Year Book Inc.:1631-51. [Google Scholar]
[2]. Woodard JR, Caldamone AA, Prune belly syndrome. In: Wein AJ (eds)Campbell Walsh Urology 2007 49th editionPhiladelphia, PASaunders Elsevier:3482-97. [Google Scholar]
[3]. Greskovich FJ 3rd, Nyberg LM Jr, The prune belly syndrome: a review of its aetiology, defects, treatment and prognosisJ Urol 1988 140:707-12. [Google Scholar]
[4]. Woodhouse CRJ, Ransley PG, Innes D, Prune Belly syndrome- report of 47 casesArchives of Disease in Childhood 1982 57:856-59. [Google Scholar]
[5]. Bargaje A, Yerger JF, Khouzami A, Jones C, Cloacal dysgenesis sequenceAnn Diagn Pathol 2008 62:6-12. [Google Scholar]
[6]. Waters EG, Cloacal dysgenesis: Related anomalies and pregnanciesObstet Gynecol 1982 59:398-402. [Google Scholar]
[7]. Escobar LF, Weavwe DD, Bixler D, Hodes ME, Mitchell M, Urorectal sptum malformation sequence: Report of six cases and embryological analysisAm J Dis Child 1987 141:1021-24. [Google Scholar]
[8]. Wright JR, Barth RF, Neff JC, Poe ET, Sucheston ME, Stempel LE, Gastrointestinal malformations associated with prune belly syndrome: three cases and a review of the literaturePaediatr Pathol 1986 5:421-28. [Google Scholar]
[9]. Quau E, Radiological Imaging of the Kidney 2010 Springer Verlag:306 [Google Scholar]
[10]. Stephens FD, Gupta D, Pathogenesis of prune belly syndromeJ Urol 1994 152:2328-31. [Google Scholar]
[11]. Terada S, Suzuki N, Uchide K, Ueno H, Akasofu K, Aetiology of prune belly syndrome: evidence of megalocytic origin in an early fetusObstet Gynecol May 1994 83:865-68. [Google Scholar]
[12]. Barsoum IB, Bingham NC, Parker KL, Jorgensen JS, Yao HH-C, Activation of the Hedgehog Pathway in the Mouse Fetal Ovary Leads to Ectopic Appearance of Fetal Leydig Cells and Female PseudohermaphroditismDevelopmental Biol 2009 329(1):96-103. [Google Scholar]
[13]. Gupta P, Kumar S, Sharma R, Gadolia A, Antenatal MRI diagnosis of cloacal dysgenesis syndromeIndian J Radiol Imaging 2010 20(2):143-46. [Google Scholar]
[14]. Patricia AB, Mac Donald EC, An epidemiological study of congenital malformation of the anterior abdominal wall in more than half a million consecutive live birthsAm J of Human Genetics 1981 33:470-78. [Google Scholar]