Introduction
Wilson’s Disease (WD), which is also known as hepatolenticular degeneration, was first defined by Dr Samuel Alexander Kinnier Wilson in 1912, [1]. Wilson’s disease (WD) is a rare, autosomal, recessive inborn error of the copper metabolism, which is caused by a mutation in the copper-transporting gene, ATP7B. The WD gene, ATP7B, is located on chromosome 13q14.3, which encodes a metal-transporting P-type adenosine triphosphatase (ATPase), which is expressed mainly in hepatocytes. The incidence of WD is estimated to be 1 in 30,000 individuals and the carrier frequency is approximately 1 in 90 [2]. An absent or a reduced function of the ATP7B protein leads to a decreased hepatocellular excretion of copper into bile. This results in hepatic copper accumulation and injury. Eventually, copper is released into the bloodstream and it is deposited in other organs, notably in the brain, kidneys, and the cornea. The hepatic production and the secretion of the ceruloplasmin protein without copper and apoceruloplasmin, result in the decreased blood level of ceruloplasmin which is found in most of the patients with WD, due to the reduced half-life of apoceruloplasmin [3]. The manifestations are more likely to be hepatic in early childhood and to be neurological in adolescents [4]. The neurological manifestations of Wilson’s disease can vary extremely and they are often diagnosed after long delays. Here, we are reporting Wilson’s disease with only neurological presentations in two siblings.
Case Report
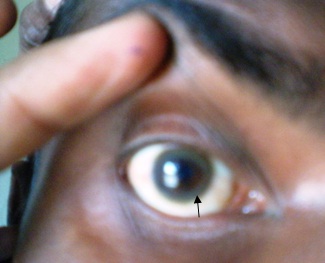
Case 1: A 14 years old Hindu boy, who was the youngest of 4 siblings, who was a product of a non – consanguineous marriage, presented with pain in right knee joint and difficulty in walking for 4 months, 10 months back, which was treated by many physician with some symptomatic improvement. After 4 months of the onset of the pain, he had developed abnormal movements of the right upper and lower limbs in the form of repetitive purposeless abnormal movements, which were progressive and which involved all the four limbs over two months of duration and for the past 4 months, he had been non – ambulatory. His parents consulted many physicians and neurologists for these problems but the diagnosis was missed. There was no improvement, and he was then brought to our institute. This abnormal movements subsided during sleep. The patient also had progressive dysarthria. His elder brother also had a history of very slowly progressive abnormal movements and dysarthria. There was no history of measles during early childhood and convulsion. His developmental milestones were normal. On examination, his vital signs were found to be normal. There was no pallor, icterus or significant lymphadenopathy. His nervous system examination revealed dystonia, exaggerated deep tendon reflexes, ankle clonus and a positive Babinski’s sign. His muscle power was >4/5 in all the limbs. The Kayser – Fleischer ring (K – F ring) was visible on both sides by the naked eye [Table/Fig-1], which was confirmed on an ophthalmoscopic examination which was done by an ophthalmologist by slit lamp examination. His other systemic examinations did not reveal any abnormality.
Kayser – Fleischer ring (K – F ring) [Arrow] due to copper deposition on descement membrane even visible on naked eye in this case
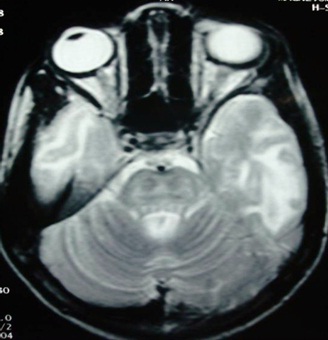
His complete blood count revealed haemoglobin 10 gm/dl, a total leukocyte count of 5600/cumm (neutrophil 62%, lymphocytes 33%, monocytes 2% and eosinophils 3%) and platelets –340,000/cumm. The serum electrolytes and the renal functions were normal. The total serum bilirubin was 0.6 mg/dl (direct bilirubin 0.2 mg/dl), the total serum protein was 6.8 gm/dl (Albumin 4.2 gm/dl) and the serum transaminases (AST, ALT) and Alkaline Phosphatise (ALP) were 38, 24, and 168 IU/L respectively. His Prothrombin Time (PT) and his activated Partial Thromboplastin Time (aPTT) were within normal limits. His serum ceruloplasmin was 95 mg/L (normal 180–350 mg/L) and his 24 hours urine copper excretion was increased to 160 ¼g (normal 24 hours urine excretion 20 – 50 ¼g). Liver biopsy was not done because his parents had not given their consents for that invasive procedure. Genetic testing for mutation analysis was not done due to financial constraints. His urine routine and microscopic examinations were normal. Ultrasonography of the abdomen was normal. Magnetic resonance imaging (MRI) of the brain, on the T2 weighted axial sequence through the pons revealed the “Face of the miniature Panda” [Table/Fig-2]. The T2 weighted image also revealed hyperintensities in the thalami and the pontine tegmentum.
T2 – weighted axial MRI sequence of the brain through pons revealed ‘face of the miniature panda’
Case 2: A 18 years old Hindu boy, the 2nd youngest of 4 siblings (the elder brother of the above mentioned patient), a product of a non-consanguineous marriage, presented with a history of progressive dysarthria for the past 5 years and a progressive, purposeless, repetitive, abnormal movement in the form of dystonia for the past 2 years. This abnormal movement subsided during sleep. He had been treated by many physicians and neurologists with antiepileptic drugs, without any improvement There was no history of measles during early childhood and convulsion. On examination, his vital signs were found to be normal. His nervous system examination revealed dysarthria, dystonia, exaggerated deep tendon reflexes, ankle clonus and a positive Babinski’s sign. His muscle power was normal. On ophthalmoscopic examination, the Kayser – Fleischer ring (K – F ring) was found to be present on both the sides. His other systemic examinations were normal. His haemogram, liver function test and renal function test and ultrasonography of the abdomen were normal. His serum ceruloplasmin was 110 mg/ L (normal 180- 350 mg/L) and his 24 hours urine copper excretion was increased (175 ¼g). His urine routine and microscopic examinations were normal.
The diagnosis of Wilson’s disease with a neurological manifestation was made on the basis of the dystonia, the K–F ring in the both eyes, the low serum ceruloplasmin, and the 24 hours urinary copper excretion. His parents were explained regarding the prognosis of the disease and the available treatment in our country. Both the patients were started on oral zinc (as zinc acetate) at a dose of 1mg/kg/dose of elemental zinc 8 hourly and on Trihexiphenidyl to control the dystonia. He was advised to avoid food with a high copper content, such as chocolates, nuts, legumes, mushrooms, shellfish and liver. His parents were also advised to avoid the use of copper utensils in the household for the storage of water and for cooking food. After two weeks of treatment, his dystonia reduced in intensity.
Discussion
Wilson’s Disease (WD) is a rare, autosomal, recessive, inborn error of the copper metabolism, which is caused by a mutation in the the copper-transporting gene, ATP7B [2]. Copper first accumulates in the liver; after the liver storage capacity for copper gets saturated, copper gets redistributed, with accumulation in the nervous system, the cornea, the kidneys and other organs [5]. Most of the patients present in the second decade of life with a primary hepatic presentation, with the remainder of the patients presenting during the third and fourth decades, with a primarily neurologic or a psychiatric presentation [6]. However, in our study, both the cases presented with neurological manifestations in the 2nd decade of life (adolescent), which was similar to the findings of Kalra et al., as was seen in their study which was done at the All India Institute of Medical Sciences (AIIMS) [4]. In most of the Indian studies, the disease was found to manifest at a younger age in Indian children. This could be most likely due to higher average intake of copper in India, which ranges from 5.7 – 7.1 mg/day and it is higher than the reported 0.34 – 1.1 mg/day in the western countries. The practice of cooking food and storing drinking water in copper/copper alloy pots might be contributory [5,7].
In Wilson’s disease with neurological presentations, the sympatomatology is predominantly extra pyramidal, like dystonia, tremors, dysphasia, dysarthria, and ataxia. The neurological symptoms are usually secondary to the cerebral copper deposition, which is sufficient to destroy the nerve cells. Both our cases had neurological manifestations, predominantly dystonia, dysarthria and some cognitive impairment. None of our patients had either a clinical nor a biochemical evidence of a hepatic involvement.
The serum ceruloplasmin levels should not be considered for making a definitive diagnosis, because they are normal in up to 10% of the affected patients and are reduced in 20% of the carriers. The Kayser – Fleischer rings can only be diagnosed definitively by an ophthalmologist by using a slit lamp.
Urine copper is an important diagnostic tool but it must be collected carefully to avoid contamination. An estimation of the 24-hour urinary copper excretion is another reliable test which can be done for the confirmation of WD. The normal excretion of copper is between 20 and 50¼g per day; in the cases of WD, the excretion is increased to in excess of 100 Normal excretion is between 20 and 50 ¼g per day; in cases of WD, excretion is increased to in excess of 100¼g per day. The “Gold standard” for the diagnosis remains a liver biopsy with quantitative copper assays. The affected patients have values of > 200¼g/gm dry weight of the liver. The copper stains are not reliable [8].
In both the cases, we had made the diagnoses on the basis of the clinical presentations, the serum ceruloplasmin levels, the 24 hours urinary copper excretion and neuroimaging (MRI). Although a liver biopsy with quantitative copper assays is the “Gold standard”, due to the parents’ refusal, liver biopsies were not done in our patients.
MRI is a very sensitive method for revealing the abnormalities in WD. On the T1 – weighted sequence, hypointensities in the basal ganglia are seen in two-thirds of the cases while in the T2 – weighted images, hyperintensities in the basal ganglia, the white matter, the thalamus or the brainstem are seen. These abnormalities are caused by a neuronal loss, gliosis, degeneration of the fibres, and vacuolisation, which are associated with the increased water content in the brain. The typical ‘face of the giant panda’ can be seen in the midbrain on the T2 – weighted axial MRI sequence of the brain and the ‘face of the miniature panda’, can be seen in the tegmentum region of the pons in the T2 – weighted sequence [9,10]. In our case, the T2 – weighted axial sequence through pons revealed the “face of the miniature panda”, which was also suggestive of Wilson’s disease.
Penicillamine was previously the primary anticopper treatment, but now, it plays only a minor role because of its toxicity and because it often worsens the existing neurologic disease if it is used as the initial therapy. If penicillamine is given, it should always be accompanied by 25 mg/d of pyridoxine. Trientine is a less toxic chelator and it supplants penicillamine when a chelator is indicated. The drug of choice for Wilson’s disease with neurologic/neuropsychiatric symptoms is Ammonium tetrathiomolybdate, but in most of the countries, it is not available commercially and it is still an experimental medicine. For the patients with hepatitis or cirrhosis, without an evidence of a hepatic decompensation or neurologic/psychiatric symptoms, zinc is the therapy of choice, although some advocate the therapy with trientine. Zinc has proven its efficacy in Wilson’s disease and it is essentially nontoxic. It produces a negative copper balance by blocking the intestinal absorption of copper, and it induces hepatic metallothionein synthesis, which sequesters the additional toxic copper. All the presymptomatic patients should be treated prophylactically, since the disease is close to 100% penetrant [8,11]. Both of our cases were treated with oral zinc (as zinc acetate) in high doses (1mg/kg/dose 8 hourly) and trihexyphenidyl 4mg 8 hourly. After 2 weeks of treatment, an improvement in the symptomatology was seen in the both our cases.
Genetic therapy and hepatocyte transplantation represent the future curative treatments for WD, along with the currently available liver transplantation [12].
Conclusion
The present cases gave us valuable information that Wilson’s disease is an uncommon, autosomal, recessive, metabolic disorder, which is often missed but easily treatable. A high index of suspicion is required while dealing with adolescents and young adults with abnormal movements and neurobehavioural abnormalities. A high degree of suspicion and an early detection of WD is critical, because an early initiation of the treatment can prevent a catastrophic outcome.
[1]. Wilson SAK, Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liverBrain 1912 34:295-509. [Google Scholar]
[2]. Huster D, Wilson’s diseaseBest Pract Res Cl Ga 2010 24(5):531-39. [Google Scholar]
[3]. Holtzman NA, Gaumnitz BM, Studies on the rate of release and turnover of ceruloplasmin and apoceruloplasmin in rat plasmaJ Biol Chem 1970 245:2354-58. [Google Scholar]
[4]. Kalra V, Wilson’s disease: early onset and lessons from a pediatric cohort in IndiaIndian Pediatr 2000 37:595-601. [Google Scholar]
[5]. Sokol RJ, Wilson’s Disease and Indian childhood cirrhosisIn: Diseases in Children. Eds. Suchy, Fredrick J 1994 747St LouisMosby Year Books Inc-72. [Google Scholar]
[6]. Walshe JM, The Liver in Wilson’s Disease (Hepatolenticular Degeneration)In: Diseases of the Liver. Eds Schiff L, Schiff ER 1982 PhiladelphiaJ. B. Lippincott:1037-84. [Google Scholar]
[7]. Ferenci P, Czlonkowska A, Stremmel W, Houwen R, Rosenberg W, Schilsky M, Jansen P, Moradpour D, Gitlin J, EASL clinical practice guidelines: Wilson’s diseaseJ Hepatol 2012 56(3):671-85. [Google Scholar]
[8]. Brewer George J, Wilson Disease. Harrison’s Principle of Internal Medicine 2008 17th EdnMc Graw Hill Medical:2449-51. [Google Scholar]
[9]. Page RA, Clinical correlation of brain MRI and MRS abnormalities in patients with Wilson diseaseNeurology 2004 63:638-43. [Google Scholar]
[10]. Kuruvilla A, Joseph S, ‘Face of the giant Panda’ sign in Wilson’s disease: revisitedNeurol India 2000 48:395-96. [Google Scholar]
[11]. Wu ZY, Lin MT, Murong SX, Wang N, Molecular diagnosis and prophylactic therapy for presymptomatic Chinese patients with Wilson diseaseArch Neurol-Chicago 2003 60(5):737-41. [Google Scholar]
[12]. Malhi H, Joseph B, Schilsky ML, Gupta S, Development of cell therapy strategies to overcome copper toxicity in the LEC rat model of Wilson diseaseRegen Med 2008 3(2):165-73. [Google Scholar]