The Sertoli Cell Only Syndrome and Glaucoma in a Sex – Determining Region Y (SRY) Positive XX Infertile Male
Manish Jain1, Veeramohan V2, Isha Chaudhary3, Ashutosh Halder4
1 Scientist, Department of Reproductive Biology, AIIMS, New Delhi, India.
2 Senior Research Fellow, Department of Reproductive Biology, AIIMS, New Delhi, India.
3 Student, Senior Research Fellow, Department of Reproductive Biology, AIIMS, New Delhi, India.
4 Additional Professor, Reproductive Biology, AIIMS, New Delhi, India.
Name, Address, E-Mail Id of The Corresponding Author: Dr Ashutosh Halder, Additional Professor, Department of Reproductive Biology, All India Institute of Medical Sciences, New Delhi 110029, India.
Phone: 91-11-26594211
E-mail: ashutoshhalder@gmail.com
The XX male syndrome is a rare genetic disorder. The phenotype is variable; it ranges from a severe impairment of the external genitalia to a normal male phenotype with infertility. It generally results from an unequal crossing over between the short arms of the sex chromosomes (X and Y). We are reporting a case of a 38-year-old man who presented with infertility and the features of hypogonadism and glaucoma. The examinations revealed normal external male genitalia, soft small testes, gynaecomastia and glaucoma. The semen analysis showed azoospermia. The serum gonadotropins were high, with low Anti Mullerian Hormone (AMH) and Inhibin B levels. The chromosomal analysis demonstrated a 46, XX karyotype. Fluorescent In-Situ Hybridization (FISH) and Polymerase Chain Reaction (PCR) revealed the presence of a Sex-determining Region Y (SRY). Testicular Fine Needle Aspiration Cytology (FNAC) revealed the Sertoli Cell Only Syndrome (SCOS). The presence of only Sertoli Cells in the testes, with glaucoma in the XX male syndrome, to our knowledge, has not been reported in the literature.
XX Male Syndrome, Sex-determining Region Y Positive, Sertoli Cell Only, Glaucoma
Case Description
The case which has been presented here is that of a 38 years old male, who was referred for non obstructive azoospermia, for its diagnosis and management and for reproductive counseling. The patient was investigated and managed in various departments viz., Reproductive Biology, Sex and Marriage Counseling Clinic, Ophthalmology, Radiology and Pathology. The study (primary testicular failure and this case is one such case) was approved by the Institutional Human Ethics Committee. He was found to have glaucoma in the left eye at presentation. The glaucoma (left eye) was diagnosed in 2001, with an increased left intraocular pressure (20.6 mm Hg) and cupping of the left optic disc. The patient was treated medically as well as surgically (by peripheral iridectomy of both the eyes). Presently, his field of vision in the left eye is severely compromised. He has been married since the past 12 years, without any issue. His clinical examination revealed no abnormality (general, local and systemic), except features of hypogonadism. His height (162 cm), weight (63 Kg), etc were normal. He was relatively short in height, in comparison to his father and brothers (175 cm – 180 cm). His external genitalia were normal (penis and scrotum) with the normal male type pubic/axillary hairs; [Table/Fig-1A]. His secondary sexual characters were normal initially; however, he complained of hypogonadism since the past 3 – 4 years (less facial hair, Tanner stage II gynaecomastia and a decreased libido; [Table/Fig-1B]. There was no varicocele or hydrocele. The volumes of the testes was 2 ml (small) and their consistency was soft. There was no history which was suggestive of a chronic persistent genital infection or of an exposure to gonadotoxins.
(Clinical Photograph) is showing well developed scrotum containing ill defined small testes
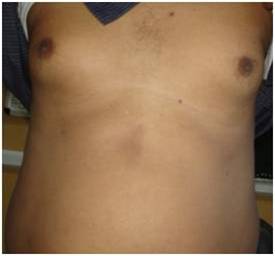
(Clinical Photograph) is showing bilateral gynaecomastia
The patient was investigated in various departments for the routine semen examination, hormones and peripheral blood chromosomes initially and later with FISH, Yq microdeletion and testicular FNAC. His semen samples were obtained by masturbation after an abstinence from sex for 3 days. The results of the semen analysis showed azoospermia (repeated). The hormonal study yielded high FSH and LH levels and low serum testosterone, AMH and Inhibin B levels. His seminal lactate level was normal [Table/Fig-2]. A conventional cytogenetics study showed a 46, XX karyotype. FISH which was done on the nuclei of the lymphocytes by using centromeric XY probes, also confirmed the XX male status and it excluded the absence of the Y (centromere) bearing cells, even at low levels (1 in 1000 cells). FISH which was done by using a SRY probe confirmed the presence of SRY on the Xp telomere [Table/Fig-3]. A Y chromosome deletion study was carried out by using 20 primer pairs for the known sequence – tagged sites (Promega, USA; Cat. no MD1531), that showed complete deletions of all the loci except the SRY gene. A Testicular FNAC which was done on both the testes showed no evidence of germ cells and it showed only the presence of the Sertoli cells [Table/Fig-4]. The patient was counseled for artificial insemination of donor (AID) or adoption, as his condition (Sertoli Cell Only Syndrome) was untreatable as of then. The patient opted for AID at a private assisted reproductive clinic and now, the couple has a baby through AID.
Showing values of reproductive endocrine/paracrine parameters
| LH miu/ml | FSH miu/ml | Testosterone ng/ml | Inhibin B pg/ml | AMH ng/ml | Seminal Lactate nmol/μl |
|---|
| Patient Value | 36.3 | 76.6 | 1.2 | 16.6 | 0.2 | 39.3 |
| Control Value | 1.5 – 12 | 1.5 – 12 | 2 to 11 | 107 – 293 | 5 – 17 | 30 – 87 |
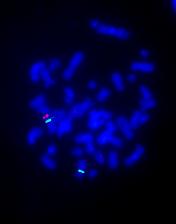
(Fluorescent In Situ Hybridization i.e., FISH) is showing triple color FISH (X centromere as green, SRY as red & Y centromere as yellow due to superimposed red with green) on a metaphase. Two green indicates 2 centromeres of X chromosome and one SRY (red) on Xpter. Absence of yellow (superimposed red & green) spot indicates no Y chromosome centromeres
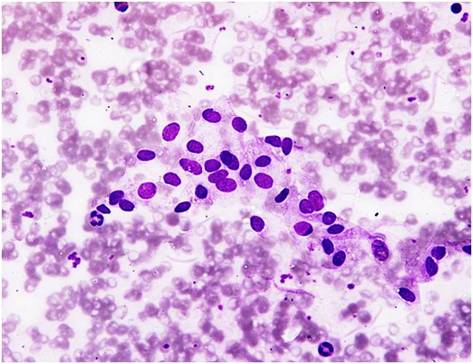
(Fine Needle Aspiration Cytology i.e., FNAC) is showing Sertoli cells aggregates in a background of leucocytes & red blood cells (smear obtained from one of the testes). There is no evidence for germ cell lineages (spermatogonia, spermatocytes, spermatids and spermatozoa)
Discussion
We have presented a case of a SRY positive, XX infertile male with the features of hypogonadism, unilateral glaucoma and SCOS. The SRY gene, in our case, was localised by the FISH study to the Xpter region. The SRY gene plays a crucial role in the testicular development and in the male sex differentiation during the early foetal life. In rare cases, this could be due to an undetected, low grade mosaicism of the XY cells. However, in our case, no mosaicism was observed, even at low levels, as the FISH analysis of the lymphocytes was negative for the Y chromosome.
The XX male syndrome presents with a spectrum of clinical appearances, from the classic XX male individuals with normal male phenotypes and infertility, to the non – classic XX male individuals with a sexual ambiguity or XX true hermaphrodites. This syndrome was first described by de la Chapelle et al., [1]. The XX male syndrome affects 1 in 20,000 of the newborn males [2]. Usually, it is caused by an unequal crossing over between the short arms (p arms) of the sex chromosomes (X and Y) during the paternal gametogenesis [3], which results in an abnormal X chromosome which contains the SRY gene. When this abnormal X combines with a normal X from the mother during fertilisation, the result is an XX male. Sometimes, the SRY autosomal translocations may cause some of the cases of the XX male syndrome [3]. The SRY gene plays a significant role in the differentiation of the testis from the undifferentiated gonad in the early embryonic life. The testis determination is initiated when the SRY gene is expressed in the pre-Sertoli cells of the undifferentiated genital ridge. The Sertoli cells play a central role in the development of a functional testis, and hence, in the expression of the male phenotype. The Sertoli cells are the first cells to differentiate in the indifferent foetal gonad, which enable the seminiferous cord formation, regression of the Mullerian ducts, prevention of the germ – cell entry into meiosis and the differentiation and the function of the Leydig’s cells. However, a subset of the XX male syndrome exists without the SRY gene [4]. In most of the classic cases, the SRY positive XX male syndrome is diagnosed in adulthood during infertility investigations. The affected males possess normal male genitalia, small testes, azoospermia and hypergonadotropic hypogonadism. The individuals with this condition sometimes have varying degrees of gynaecomastia, poor facial hair growth and a diminished libido.
The testicular histology or the cytology which are reported with this syndrome are inconsistent; some have been reported as hyalinization of the seminiferous tubules, absence of the germ cells or absence of the seminiferous tubules [5], diffuse hyalinized seminiferous tubules with Sertoli cells and marked interstitial cell hyperplasia [6], immature testes [7], Leydig’s cell hyperplasia or absence of the Leydig’s cells.
Clinically, the XX male syndrome is somehow similar to the Klinefelter’s syndrome viz., small testes, elevated gonadotropins, and low to normal testosterone levels. However, it differs with the Klinefelter’s syndrome in the height of the patients. The XX males have been reported to be significantly shorter than the Klinefelter’s patients or than healthy men, as was our case, who was shorter than his father and brothers. The short stature in the XX male syndrome, in comparison to the stature in Klinefelter’s patients or in normal men may be due to the absence of the Y – chromosome genes which are related to growth [8].
The rare findings in our case were the Sertoli Cell Only Syndrome and glaucoma. A complete Sertoli Cell – Only Syndrome has not been reported earlier. The low levels (although they were detectable) of AMH and Inhibin B in our case, provided further evidence of the presence of few and/or dysfunctional, mature Sertoli cells. The literature on the testicular biopsies of the XX males have revealed a complete lack of spermato-genic cells, with presence of only the Leydig’s and the Sertoli cells [9]. The SCOS with the combinations of the Leydig’s cells hyperplasia/hypoplasia or hyalinization of the tubules have been found in few other studies [6,10]. Some studies had also reported the presence of spermatogonia at birth in the XX male patients before puberty, but they had disappeared [11] after puberty, as was seen in Klinefelter’s syndrome also. The lack of the post pubertal germ cells explained the findings of the soft and small testes which are often associated with the XX male, as it was in our case.
The angle closure glaucoma which was present in our case has not been reported in the literature in XX males, although studies have reported an association of glaucoma with the Klinefelter’s/Variant Klinefelter’s Syndrome [12, 13]. The association of glaucoma in our case may have shared a common aetiopathogenic basis as it had shared with the Klinefelter’s syndrome, but it could also have been coincidental.
[1]. De la Chapelle A, Hortling H, Niemi M, XX sex chromosomes in a human male. First caseActa Med Scand 1964 175(Suppl 412):25-28. [Google Scholar]
[2]. Nielsen J, Sillesen I, Incidence of chromosome aberrations among 11,148 newborn childrenHumangenetik 1975 30:1-12. [Google Scholar]
[3]. Dauwerse JG, Hansson KB, Brouwers AA, An XX male with the sex-determining region Y gene inserted in the long arm of chromossome 16Fertil Steril 2006 86(463):e2-e5. [Google Scholar]
[4]. Valetto A, Bertini V, Rapalini E, 46, XX SRY-negative man with complete virilization and infertility as the main anomalyFertil Steril 2005 83:216-19. [Google Scholar]
[5]. Yumura Y, Kanno H, Ogawa T, Two cases of 46,XX male with chief complaint of infertilityHinyokika Kiyo 2003 49:727-34. [Google Scholar]
[6]. Li JH, Huang TH, Jiang XW, 46, XX male sex reversal syndromeAsian J Androl 2004 6:165-67. [Google Scholar]
[7]. Kolon TF, Ferrer FA, McKenna PH, Clinical and molecular analysis of XX sex reversed patientsJ Urol 1998 160:1169-72.discussion 1178 [Google Scholar]
[8]. Kirsch S, Weiss B, Zumbach K, Molecular and evolutionary analysis of the growth-controlling region on the human Y chromosomeHum Genet 2004 114:173-81. [Google Scholar]
[9]. Margarit E, Soler A, Carrio A, Molecular, cytogenetic, and clinical characterization of six XX males including one prenatal diagnosisJ Med Genet 1998 35:727-30. [Google Scholar]
[10]. Grigorescu-Sido A, Heinrich U, Grigorescu-Sido P, Three new 46, XX male patients: a clinical cytogenetic and molecular analysisJ Pediatr Endocrinol Metab 2005 18:197-203. [Google Scholar]
[11]. Kusz K, Kotecki M, Wojda A, Incomplete mascularization of XX subjects carrying the SRY gene on an inactive X chromosomeJ Med Genet 1999 36:452-56. [Google Scholar]
[12]. Muniesa Royo MJ, Sánchez Pérez C, Jurjo Campo C, Juvenile glaucoma and optic disc pit with macular detachment in Klinefelter’s syndromeArch Soc Esp Opthalmol 2012 87:256-59. [Google Scholar]
[13]. Pamuk BO, Torun AN, Kulaksizoglu M, 49, XXXXY Syndrome with Autoimmune Diabetes and Ocular ManifestationsMed Princ Pract 2009 18:482-85. [Google Scholar]