INTRODUCTION
Pheochromocytomas and paragangliomas are rare and they occur in approximately 0.05%-0.1% of the patients with sustained hypertension. However, this probably accounts for only 50% of the people who harbour pheochromocytomas or paragangliomas, because approximately half of the patients with pheochromocytomas or paragangliomas have paroxysmal hypertension or normotension [1].
Malignant hypertension, which is a complication of hypertension, which is characterized by an elevated blood pressure (200mm/140mm Hg), is considered as a medical emergency and it is rarely secondary to a paraganglioma. Malignant hypertension is unique in its relationship with a catecholamine secreting paraganglioma [2].
CASE HISTORY
First Case: A 12 years old boy presented with a difficulty in swallowing, severe headache and sweating. He had no other symptoms or a past medical history to note. His routine blood tests were unremarkable. His physical examination showed an elevation of his blood pressure (up to 200/150 mmHg) and slight tachycardia (90/min).
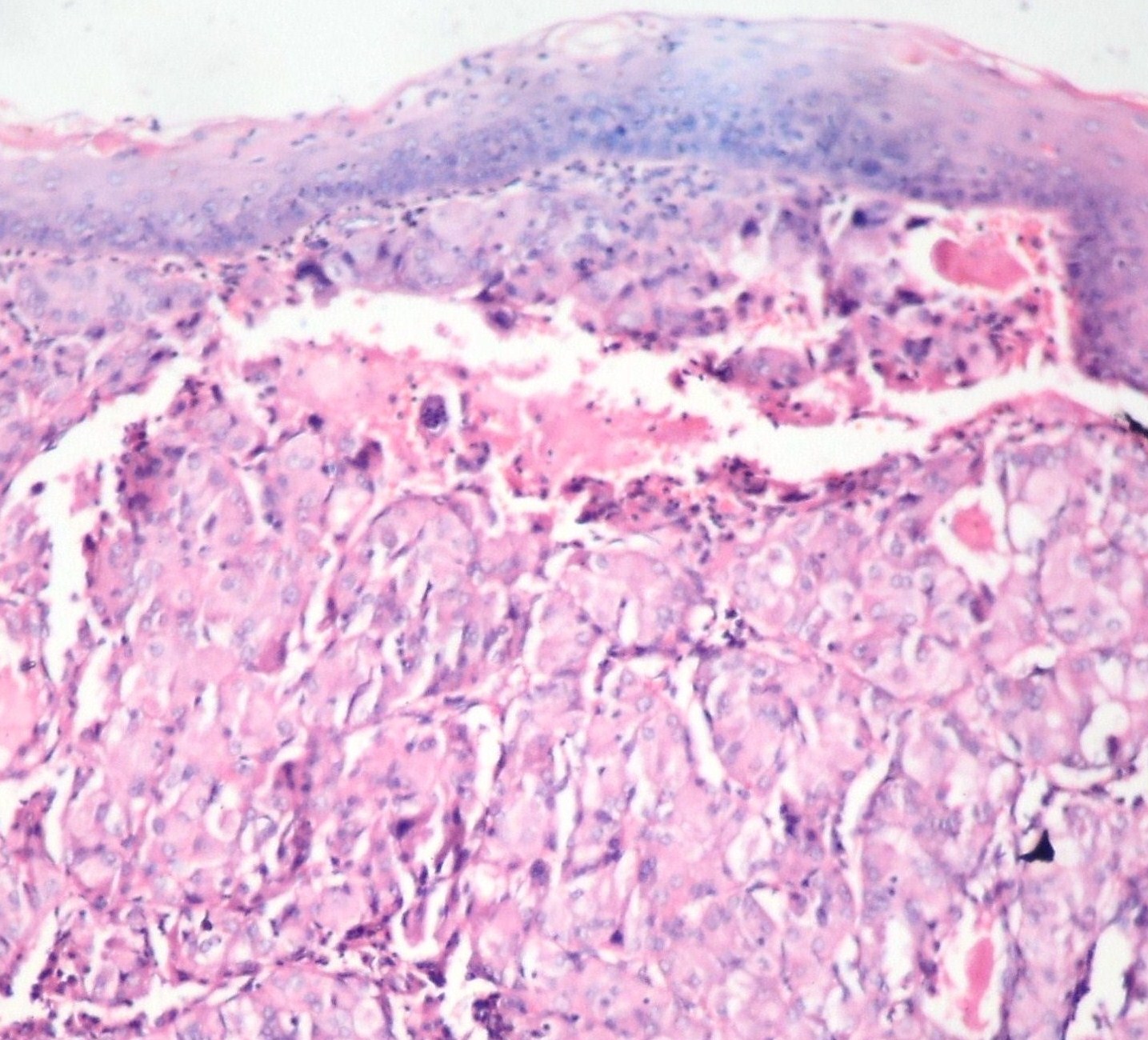
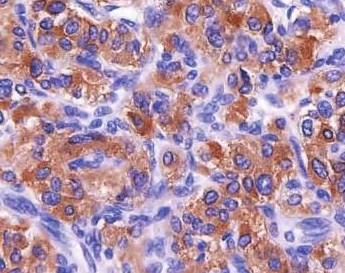
On examination, his left sided tonsil was found to be enlarged and it was smooth, without any surface ulceration. He underwent a tonsillectomy. The mass was removed. The patient died intra-operatively. The histological examination of the tonsil revealed a paraganglioma [Table/Fig-1]. It was later confirmed by the positivities that it showed for synaptophysin [Table/Fig-2] and chromogranin.
Tonsillar squamous epithelial lining with underlying tumour tissue (Low Power)
Tumour cells showing synaptophysin positivity on immunohistochemistry
Second Case: A 55 year old female presented initially with a history of frank haematuria. She had no other symptoms or a past medical history to note. Her routine blood tests were unremarkable. The 24-hours urinary vanillyl mandelic acid level was within normal limits. Her physical examination showed an elevation of her blood pressure (up to 210/140 mmHg) and slight tachycardia (90/min), which exacerbated especially after micturition.
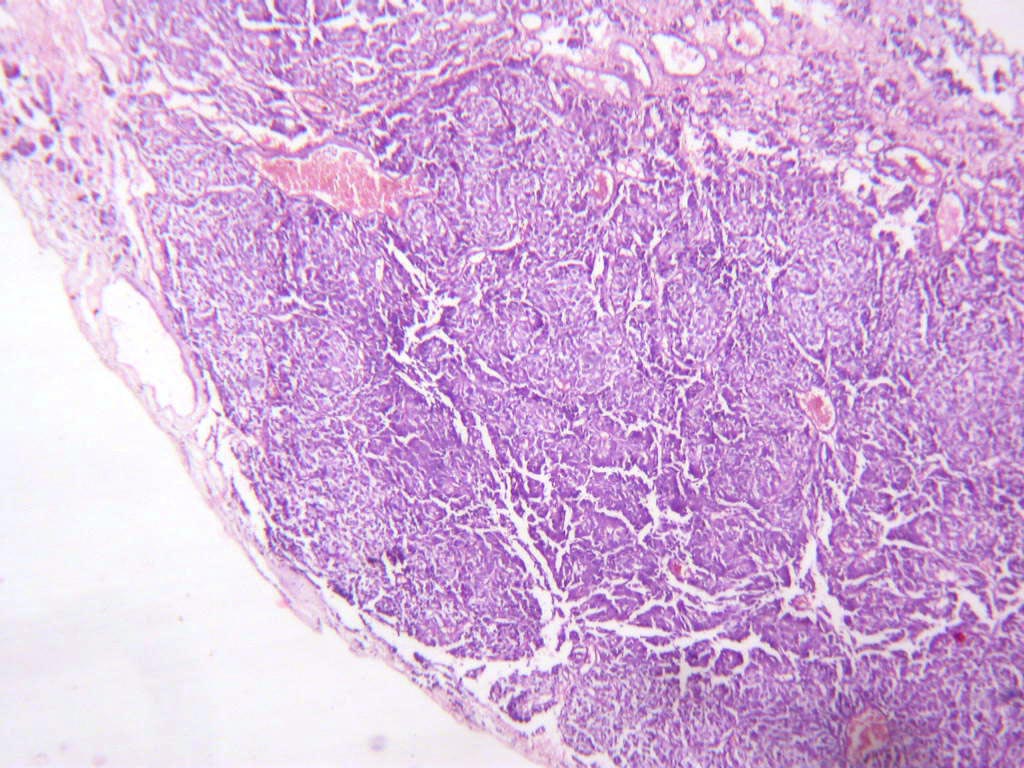
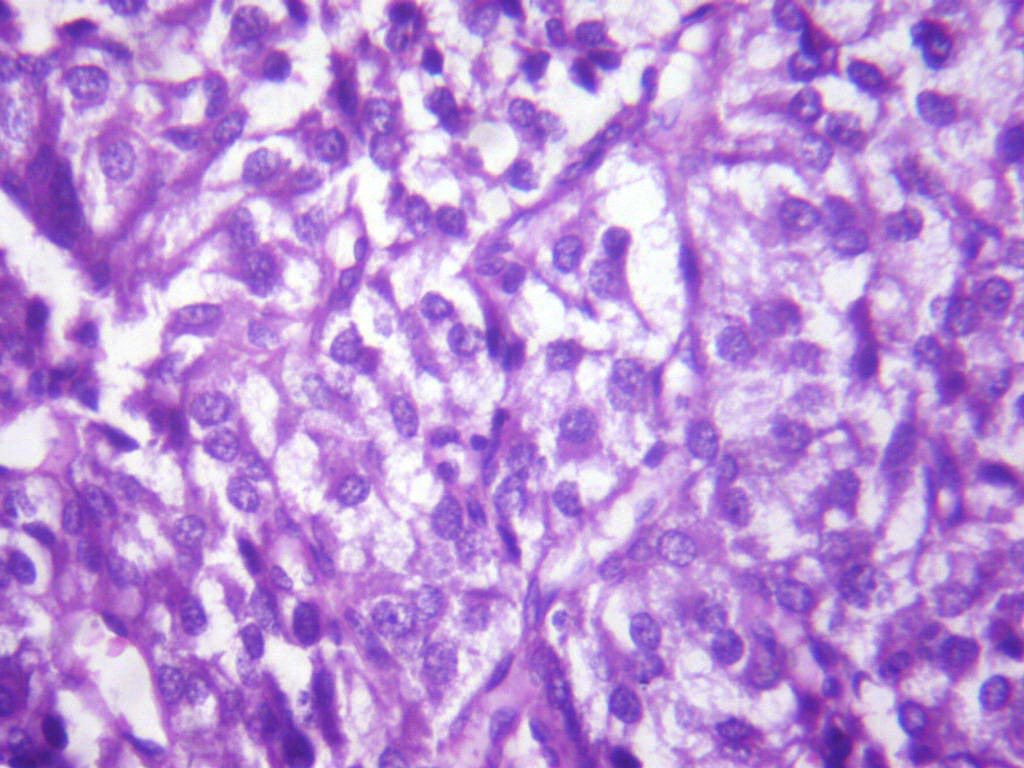
Cystoscopy revealed a smooth, well vascularized, sub mucosal mass on the dome of the bladder, without signs of a surface ulceration. She underwent a transurethral resection of the bladder tumour (TURBT). A histological examination revealed a paraganglioma [Table/Fig-3 & 4]. On doing an immunohistochemical analysis, the tumour cells were found to be positive for synaptophysin and chromogranin. This patient is under follow up and is keeping fine.
Bladder tissue showing tumour cells arranged in Zellbellen Pattern (Low Power)
Bladder tissue showing tumour cells with salt pepper chromatin (High Power)
DISCUSSION
A paraganglioma is probably the most fascinating of all the tumours, as it can present with a wide range of clinical manifestations [3]. The extra-adrenal phaeochromocytomas are known as paragangliomas. A majority of the extra-adrenal tumours occur intra-abdominally (85% occur below the diaphragm), along the sympathetic chain or from the organ of Zuckerkandl [4]. A paraganglioma of the tonsil has been reported only once in the literature [2]. Paragangliomas of the urinary bladder constitute less than 1% of all the bladder tumours and 6% of all the paragangliomas [5].
Paragangliomas usually arise in the sixth decade of life and they often present as painless masses. A paraganglioma arises from the cells of neural crest embryonically and it belongs to the amineprecursor-uptake decarboxylation system. The malignancy and the recurrence rates of this tumour are approximately 5% and 18%, respectively [4].
Almost all pheochromocytomas and sympathetic extra-adrenal paragangliomas produce, store, metabolize, and secrete catecholamines or their metabolites. Recent studies have found that approximately 20% of the head and neck paragangliomas also produce significant amounts of catecholamines [6].
The main signs and symptoms of a catecholamine excess include hypertension, palpitations, headache, sweating, and pallor. The less common signs and symptoms are fatigue, nausea, weight loss, constipation, flushing, and fever. According to the degree of the catecholamine excess, the patients may present with myocardial infarction, arrhythmia, stroke, or other vascular manifestations (eg, any organ ischaemia). Similar signs and symptoms are produced by numerous other clinical conditions, and therefore, a pheochromocytoma is often referred to as the “great mimic” [1].
In any case of a sustained, paroxysmal hypertension or a paradoxical hypertension despite the antihypertensive therapy, especially during a therapy with s-blockers, the diagnosis of a pheochromocytoma has to be kept in mind and it has to be ruled out. A new onset of hypertension while the patient is under a tricyclic antidepressive medication and a severe symptomatic hypotension when a therapy is started with α-blockers or severe retinopathy in a newly-diagnosed hypertension case may suggest a pheochromocytoma. Other forms of secondary hypertension such as renal artery stenosis, hypercortisolism and hyperaldosteronism, should be considered as the differential diagnosis. The symptoms of a pheochromocytoma can further be mimicked by hyperthyroidism, panic attacks, hypoglycaemia and alcohol withdrawal symptoms. The sudden cessation of a clonidine of the beta blocker therapy may also cause similar symptoms [7].
Malignant hypertension is a medical emergency which usually occurs secondary to uncontrolled hypertension. The rare causes of malignant hypertension include pheochromocytomas and paragangliomas. Hypertension, headache, palpitations and sweating may occur; however, a functional hormone secretion is uncommon when the tumour arises in the head and neck region (2%) [2].
Zhou M et al., in his study on 15 cases of paragangliomas, observed the following features-. histologically, the “Zellballen” and the diffuse patterns were present in 12 (80%) and 3 (20%) of the cases. The other patterns included irregular nests and pseudorosette formations.
Tumour necrosis, a significant cautery artifact, and a muscularis propria invasion were present in 1 (7%), 3 (20%) cases, and 10 (67%) cases, respectively. All the 15 tumours were composed of large polygonal cells with abundant granular cytoplasm. Focal clear cells were present in 3 (20%) cases. The nuclei were mostly uniform, although occasional pleomorphic nuclei were seen in 6 (40%) cases, and 2 (13%) cases had frequent pleomorphic nuclei. The mitoses were rare overall, and no abnormal mitotic figures were found [8].
The major histologic features that usually lead to a misdiagnosis include a diffuse growth pattern, focal clear cells, necrosis, and a muscularis propria invasion, with a significant cautery artifact compounding the diagnostic problems [8]. The immunohistochemical markers like chromogranin, neuron-specific enolase and S100 are positive in the paragangliomas. MIB1 and p53 have been studied to assess their malignant potential [5, 6].
To date, germline mutations in five genes have been described, which lead to several familial disorders which are associated with pheochromocytomas. An activating mutation of the RET proto-oncogene, which codes for a tyrosine kinase receptor, that transduces the signals which are associated with growth and differentiation, leads to MEN [2]. An abnormal VHL gene, a tumour suppressor gene, is responsible for VHL. Mutations of the neurofibromatosis type 1 gene (NF1) cause von Recklinghausen’s disease and the hereditary pheochromocytoma paragangliomas syndrome is associated with the mutations in the succinate dehydrogenase (SDH) subunit genes, SDHB and SDHD, which comprise portions of the mitochondrial complex II. The pheochromocytomas in the MEN [2], VHL and the NF1 disorders usually are not the first clinical manifestations and they are more likely to be benign and bilateral [7].
CONCLUSION
These cases are being presented because in our first case, we lost the patient, because a clinical diagnosis of a paraganglioma was not thought of, in a patient with severe headache and malignant hypertension. In the second case, the histologic features of a paraganglioma of the bladder could have been misdiagnosed as a conventional urothelial cancer in the bladder. Therefore, a clinical correlation, with a careful search for the characteristic histologic features and, if necessary, supportive immunohistochemical studies, can lead to an early diagnosis and treatment.
[1]. Chen H, The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors. Pheochromocytoma, Paraganglioma, and Medullary Thyroid CancerPancreas 2010 39(6):775-83. [Google Scholar]
[2]. Ansari MS, Goel Apul, Goel Saroj, Durairajan LN, Seth Amlesh, Malignant Paraganglioma of the urinary bladder. A Case reportInternational Urology and Nephrology 2001 33(2):343-45. [Google Scholar]
[3]. Yakoob Shahid, Malignant hypertension association with paraganglioma of the tonsilChest 2003 124(4):313-314S. [Google Scholar]
[4]. McKenzie Paraganglioma of the Bladder-A case report and reviewThe Internet Journal of Urology 2009 6(1) [Google Scholar]
[5]. Chao-Jung Wei, Malignant paraganglioma of bladder: A case reportChin J Radiol 2001 26:233-36. [Google Scholar]
[6]. Van Duinen N, Steenvoorden D, Kema IP, Increased urinary excretion of 3-methoxytyramine in patients with head and neck paragangliomasJ Clin Endocrinol Metab 2010 95:209-14. [Google Scholar]
[7]. Reisch Nicole, Peczkowska Mariola, Januszewicz Andrzej, Neumann Hartmut PH, Pheochromocytoma: presentation, diagnosis and treatmentJournal of Hypertension 2006 24:2331-39. [Google Scholar]
[8]. Shah VB, Singhal S, Pathak HR, Puranik GV, Bombay Hospital Journal 2009 51(1):132-34. [Google Scholar]