INTRODUCTION
Chronic Obstructive Pulmonary Disease (COPD) is characterized by a poorly reversible airflow limitation that is usually progressive and associated with the abnormal inflammatory response of the lung to noxious particles and/ or gases which are present in cigarette smoke [1, 2]. The characteristic features of the disease are chronic inflammation of the peripheral airways, chronic bronchitis and destruction of the lung parenchyma (emphysema), which include systemic extra-pulmonary manifestations [3]. The patients can suffer from one, some or all of these conditions. The peripheral airways inflammation or small-airway disease involves various morphological abnormalities such as airway narrowing with goblet cell hyperplasia, smooth muscle hypertrophy, excess mucous, oedema and inflammatory cellular infiltration. Airway remodeling with sub-epithelial and peribronchial fibrosis has been postulated as the critical factor in the small-airway narrowing and the fixed airway obstruction in the small-airways of the patients with COPD [4].
Anatomically, the disease can be viewed as a part of a spectrum, with chronic bronchitis at one end and emphysema at the other. Chronic bronchitis is characterized by cough and sputum production that results from the cigarette smoke, which induces mucous gland enlargement and goblet cell hyperplasia in the central airways, when the other pulmonary or cardiac causes for the chronic productive cough have been excluded [1, 5]. This inflammation is associated with increased mucous production, decreased mucociliary clearance and increased permeability of the epithelial barrier of the airways. On the other hand, emphysema is Aggarwrwaldefined, morphologically, as a permanent destructive enlargement of the peripheral airspaces of the lungs, without any obvious fibrosis, which includes the respiratory bronchioles, the alveolar ducts and the alveoli which are distal to the terminal bronchioles, which is accompanied by the destruction of the walls of these structures. The centrilobular emphysema is more closely associated with cigarette smoking [6].
The deterioration of COPD is accelerated after the acute exacerbations that vary in frequency, but ultimately culminate in severe COPD. The acute exacerbations increase the morbidity and mortality and represent a major health care burden with financial implications [7]. There is no standardized definition for an acute exacerbation, but the most common symptoms include increased breathlessness, cough, sputum production and purulence [8]. In recent years, COPD has been postulated as a systemic illness and/or as being associated with other smoking-related systemic diseases with manifestations from the organ systems other than lungs and the airways.
THE EPIDEMIOLOGY AND AETIOLOGY OF COPD
COPD is a leading cause of morbidity and mortality throughout the world, being responsible for significant disability and an increasing economic and social burden. COPD is the 4th leading cause of death worldwide. The incidence of the disease is increasing year by year and it has been estimated that by 2020, COPD will be the 3rd most common cause of death and the 5th most common cause of global disability [1, 9]. The COPD mortality in females has increased significantly in the last 20 years. There are striking differences between the prevalence of COPD in various countries and between the sexes, even when identical detection methods are used [10, 11]. Recent studies have shown that significant numbers of patients with COPD and chronic bronchitis can be detected, even among young smokers who have a 10-years smoking history [12]. Increasing attention is being focused on COPD, even in the developing countries [13]. In India, several regional surveys have shown impressive data [14], although the exact incidence and the prevalence of COPD are unknown. The prevalence of the disease is higher in ‘bidi’ smokers as compared to that in ‘cigarette’ smokers (8.2% versus 5.9% respectively) [13, 14]. However, the total burden of COPD is underestimated, since no significant clinical symptoms are experienced in the early stages of the disease
THE RISK FACTORS IN COPD
The most important factor which causes COPD is smoking, which is the modern pandemic which draws heavy tolls in terms of human suffering and the health economics. The annual mortality which is attributable to tobacco smoking will increase to 3 million by 2025 in the developed world and to 7 million in the developing world, if the smoking habits continue [15]. It has been reported that only 10-20% of the heavy smokers develop an irreversible airway limitation, thus suggesting that other environmental or genetic factors may contribute to COPD [9,16], but recent studies have observed that up to 50% of the smokers might develop the disease [17] and that this number may increase even more when different environmental factors i.e. pollen, animal dander, other inhaled irritants than cigarette smoke and cold air are involved [18]. The outdoor and indoor air pollution from an urban environment provides a common ground to respiratory diseases such as asthma, allergy and COPD.
Thus, smoking has resulted in a ‘smoky grey’ plague which can replace the ‘white’ plague of the tuberculosis epidemic, largely because of the increasing smoking habits and an incomplete understanding of the pathobiology of COPD. Besides, certain specific genetic factors which have been discussed below.
The aetiology of COPD involves a complex interplay between the acquired primary risk factors such as tobacco smoke, environmental pollutants, infections and occupation on one hand and the predisposing risk factors (alone or in combination) such as the airway responsiveness/reactivity, diet, hygiene, antioxidant defense potential, gender and age on the other [19] [Table/Fig-1].
Risk Factors Associated with Copd.
(Genetic deficiency of α1-antitrypsin, in the presence of other risk factors, enhances the probability of developing COPD). ADS: Antioxidant defense Systems; AHR: Airway hyper- responsiveness/reactivity.
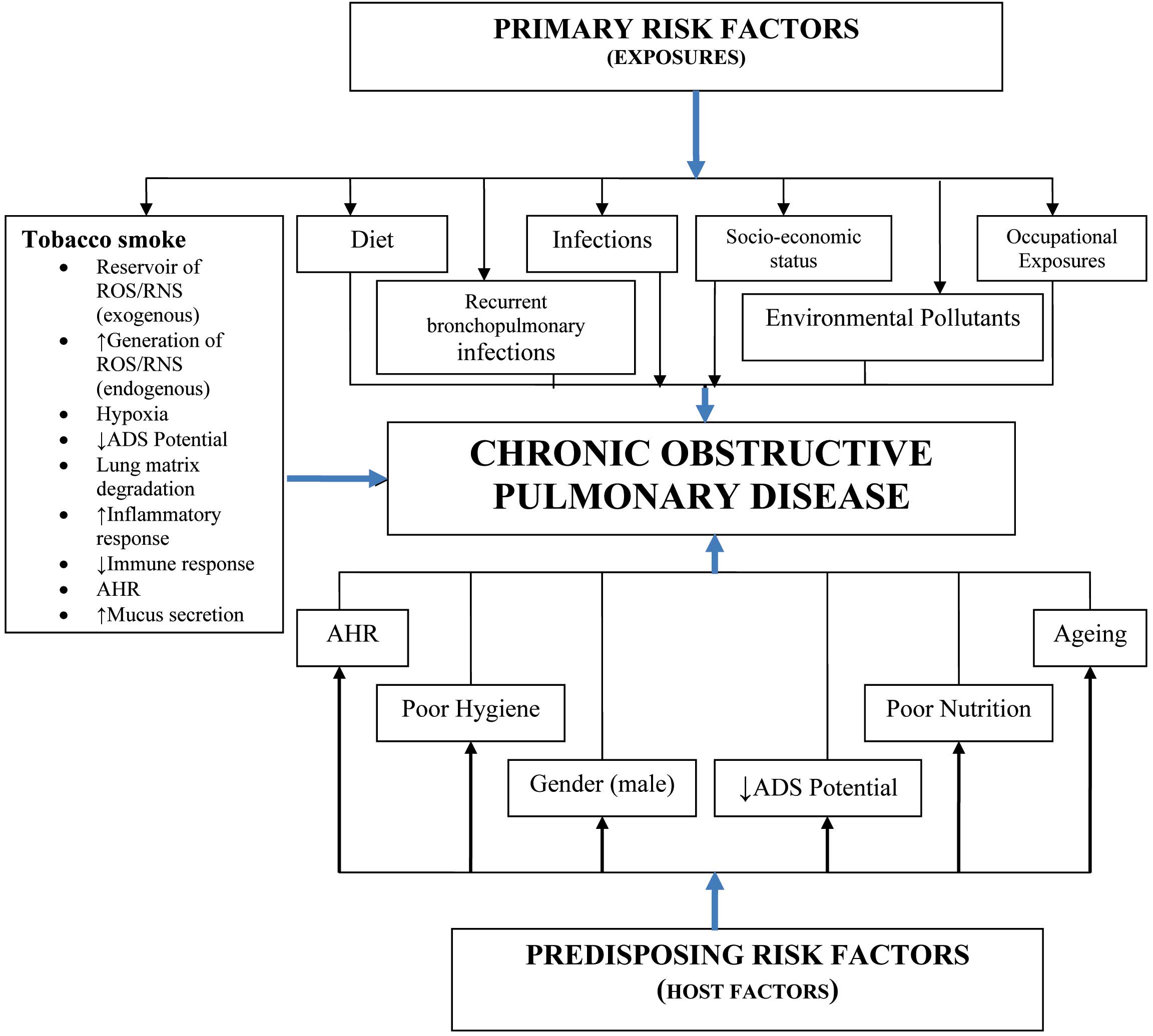
Although smoking is the major risk factor in the development of COPD [20], only 15-20% of all the smokers develop airway obstruction and therefore not all smokers acquire COPD, and non-smokers can also develop COPD [21, 22]. A population survey which was carried out in India documented that smokers were at a 2.5-times higher risk for developing COPD as compared to the non-smokers [12]. Understanding why this sub-population is susceptible to the lung injuries which result from the use of tobacco smoke, remains elusive. In COPD, as in many other clinical conditions, a complete description of the molecular components that encode a particular phenotype, would not allow for a complete understanding of the disease, without taking into account the influence of other risk factors. COPD, like asthma, seems to be a multi-genetic disease, and in the literature, there are many reports which pinpoint a specific genetic determinant of the airway function as well as a predisposing factor that may confer susceptibility to COPD [23]. Studies which have been done on monozygotic twins have suggested a genetic predisposition to COPD, although the exact mode of transmission, if any, remains to be established [24]. Both experimental as well as clinical studies have shown an association between an alpha1-antitrypsin (α1-AT) deficiency and COPD. The α1-AT is an acute phase reactant that plays a critical role in the pulmonary defense system [25]. The reactive centre of α1-AT fits into the active site of neutrophil elastase, thus forming a tight complex which is then removed from the circulation by degradation. Thus, the exposed reactive centre of α1-AT acts as a ‘bait’ for elastase and it is known as a molecular ‘mousetrap’. This phenomenon is also seen with other anti proteases such as serum α2-macroglobulin and the secretory leukoprotease inhibitor which is present in the bronchial mucus [26].
As far as the α1-AT deficiency is concerned, the Z allele is the commonest deficient variant due to a point mutation, Glu342-to-Lys, which is associated with a steep decline in the lung function. The other abnormalities range from non-functional proteins (M variant), increased catabolism (S variant), spontaneous polymerization and reduced secretion (Z variant), to absence of the product (null variants) [26]. Besides the α1-AT variants, the polymorphisms in several other genes have also been recently linked to the development of COPD, e.g. tumour necrosis factor-α promoter [26], glutathione-S-transferase [23], extracellular superoxide dismutase [24], dopamine receptors [27], epoxide hydrolase [28], matrix metalloproteinases (MMPs) [29], p53 [30], haemoxygenase [28], cytochrome P450 [28], etc.
THE BIOCHEMICAL BASIS OF THE OXIDATIVE/ NITROSSIVE STRESS IN COPD
ROS/RNS are produced as the by-products of the normal meta-bolic processes in all aerobic organisms. Many ROS/RNS are the result of the naturally occurring processes such as oxygen metabolism and inflammatory processes. For example, when the cells use oxygen to generate energy, free radicals are created as a consequence of the production of ATP by the mitochondria. Exercise can increase the levels of the free radicals, as can environmental stimuli such as ionization radiation (from industries, sun exposure, cosmic rays, and medical X-rays), environmental toxins, altered atmospheric conditions (e.g. hypoxia and hyperoxia), ozone and nitrous oxide (primarily from automobile exhaust). Lifestyle stressors such as smoking and excessive alcohol consumption are also known to affect the levels of the free radicals. The radical species may combine to form other more damaging or toxic species such as hydroxyl free radical and peroxynitrite (a product of the reaction between the superoxide and nitric oxide radicals). In physiological conditions, the Antioxidant Defense Systems (ADS) in the body protect the cells and tissues against these deleterious species [31].
Nearly every organ system can be found to have a basic degree of oxidative/nitrosative “Achilles heel”. With the current understanding that free radicals can act as cell signaling or messenger agents, it is likely that they may also play a role in the normal function as well as in various disease aetiologies. When the generation of ROS/RNS exceeds the ability of ADS to neutralize and eliminate them, such an imbalance can cause oxidative/nitrosative stress and it results in a damage to the cellular constituents (phospholipids, proteins, lipoproteins, nucleic acids and sugar) [31]. Because of this, oxidative/nitrosative stress has been implicated in cardiovascular diseases such as atherosclerosis, ischaemia/reperfusion injury, restenosis and hypertension; in cancer, in inflammatory diseases such as Acute Respiratory Distress Syndrome (ARDS), asthma, Inflammatory Bowel Disease (IBD), dermal and ocular inflammation and arthritis; in metabolic diseases, such as diabetes; in diseases of the Central Nervous System (CNS), such as Amyotrophic Lateral Sclerosis (ALS), Alzheimer’s disease, Parkinson’s disease and stroke, as well as in the aging processes [32, 33].
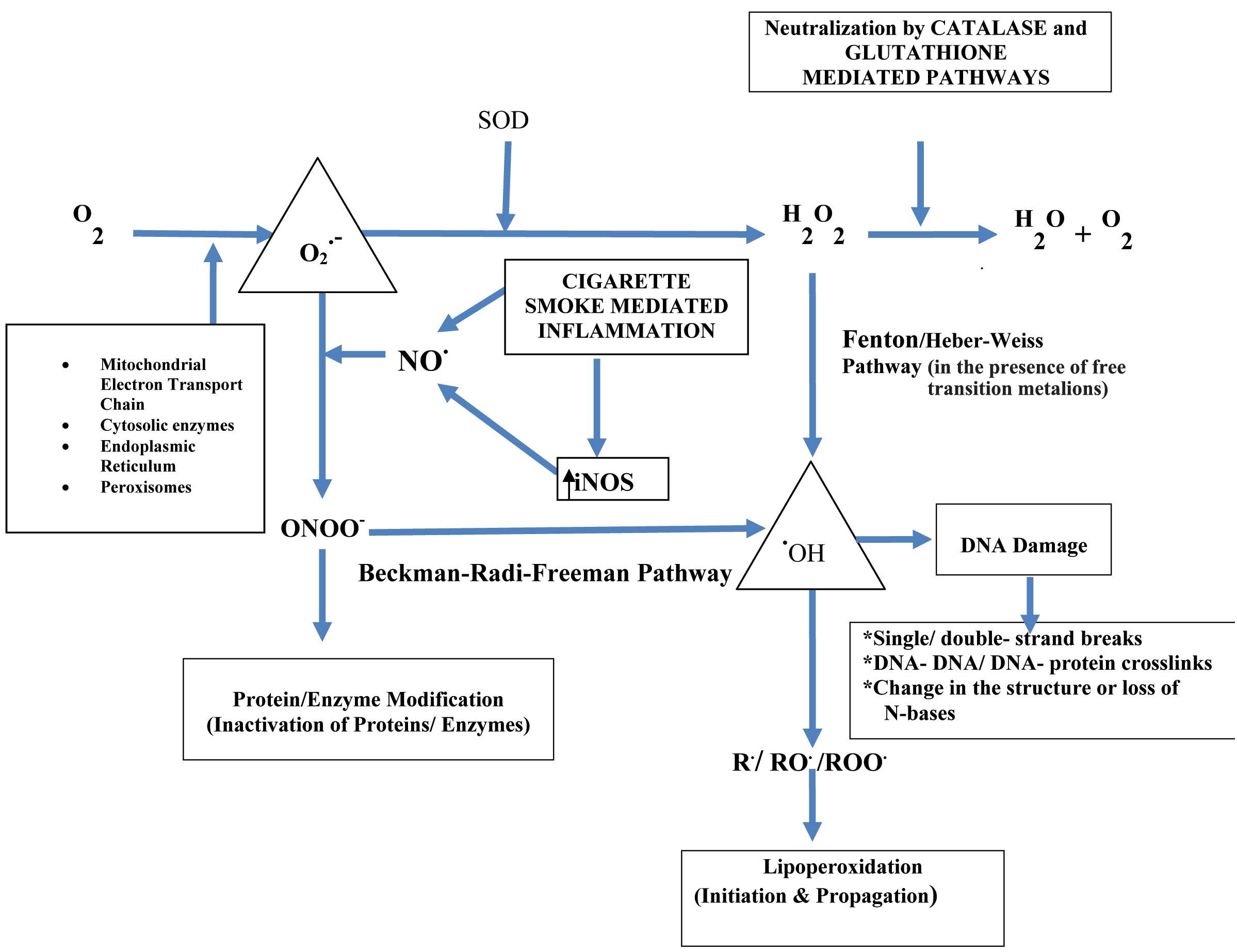
Cigarette smoke, a cocktail of toxic chemicals which include ROS/RNS, contains 10 [17]-10 [20] oxidant molecules/puff, of which nearly 10 [14] are oxygen free radicals in the gas phase alone, particularly high levels of nitric oxide. The tar phase has an equally abundant number of ROS and RNS, which include phenols and quinine [30] [Table/Fig-2]. In COPD, oxidative/nitrosative stress occurs in the small airways, the lung parenchyma and the alveolar regions, and it is associated with the activation of the cytokines and growth factors and the activated inflammatory cells produce large amounts of ROS/RNS. Most of the ROS/RNS which are produced in the lung tissue come from neutrophils, alveolar macrophages and eosinophils, but also the bronchial, the alveolar epithelial and the endothelial cells are capable of producing ROS/RNS due to inflammation. These radicals which are either inhaled (cigarette smoke) or produced endogenously, are chemically reactive molecules.
Tobacco Smoke: A Reservoir of ROSs/RNSs
| Reactive Oxygen Species (ROSs) |
| Oxygen radicals | Non-radicals |
| O2.- Superoxide Anion | ONOO- Peroxynitrite |
| OH. Hydroxyl radical | H2O2 Hydrogen peroxide |
| R. Alkyl radical | |
| RO. Alkoxyl radical | |
| ROO. Peroxyl radical | |
| HQ. Semiquinone radical | |
| Reactive Nitrogen Species (RNSs) |
| Nitrogen radicals | Non-radicals |
| NO. Nitric oxide radical | ONOO- Peroxynitrite |
| NO2. Nitrogen dioxide radical | |
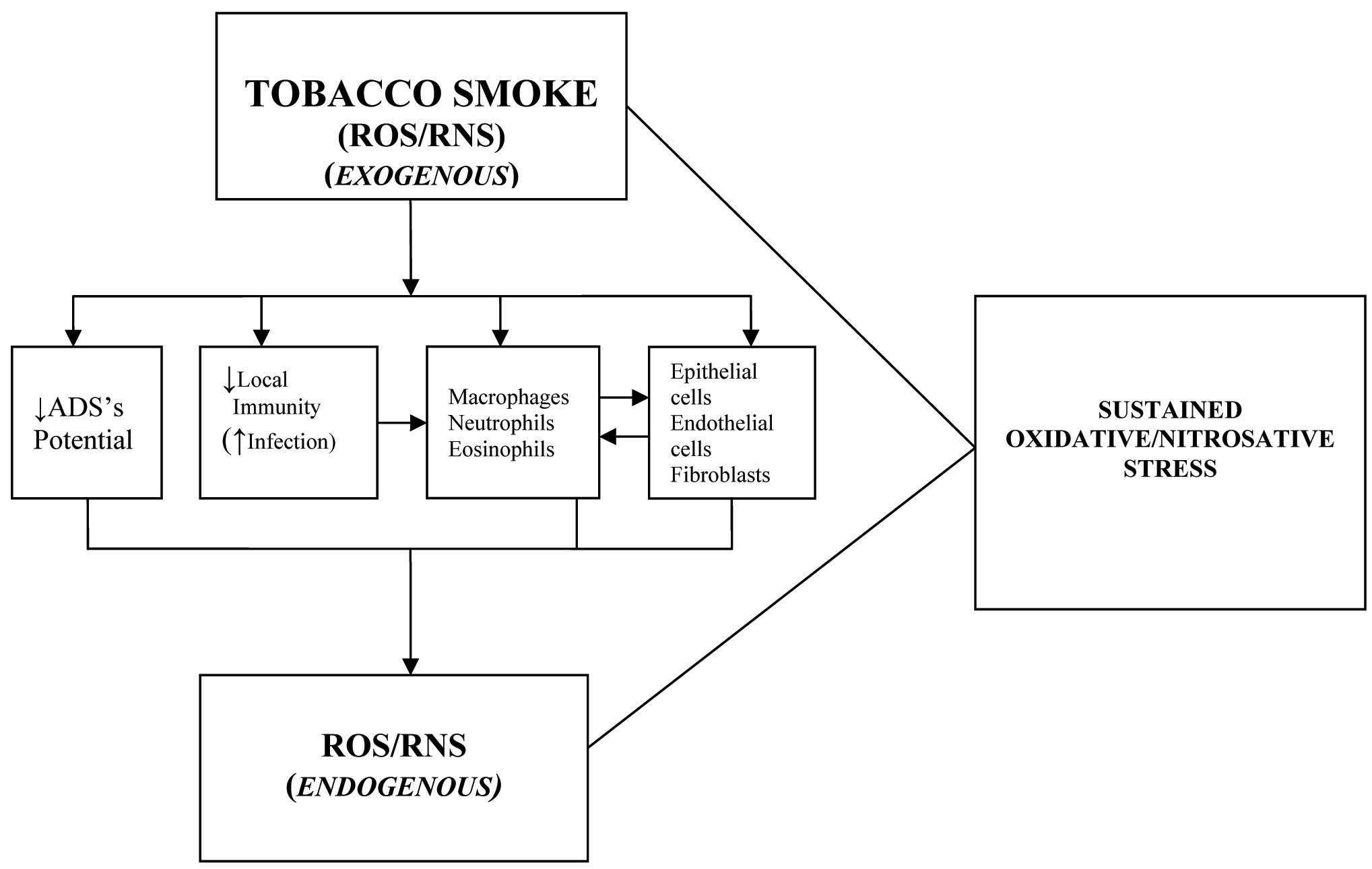
The smokers harbour a higher number of alveolar macrophages which release ROS and neutrophil chemotactic factors [34]. The neutrophils are the archetypical inflammatory cells which have been primed to release ROS, lysosomal enzymes and lipid and protein mediators; although these molecules can also be derived from other cells, e.g. macrophages, epithelial cells, etc [35]. The major producers of the superoxide radicals include enzymes and their breakdown can be spontaneous and enzymatic. The generation of oxidants is further enhanced by the presence of free transition metals (Fe2+ and Cu2+) in the airspaces, which are released due to the damage which is done to the cellular metal-binding proteins [36]. As a result of this, ‘Fenton’ and ‘Haber-Weiss’ reactions occur, generating highly reactive hydroxyl free radicals (OH.) which are particularly deleterious to the biological membranes [Table/Fig-3]. The endogenous release of ROS/RNS as a part of the host response, culminates in a sustained oxidative/nitrosative stress in chronic smokers [37] [Table/Fig-4].
Sources of ROS/RNS Generated Endogenously by Pulmonary Cells.
O2: oxygen; O2.- : superoxide anion radical; H2O2: hydrogen peroxide; .OH: hydroxyl radical; NO: nitric oxide; ONOO-: peroxynitrite; SOD: superoxide dismutase; H2O:water; iNOS: inducible nitric oxide synthase; R. : Alkyl radical; RO. :Alkoxyl radical and ROO. :Peroxyl radical
The Link between Tobacco Smoke and Oxidative/Nitrosative Stress
ROS: Reactive Oxygen Species; RNS: Reactive Nitrogen Species; ADS: Antioxidant defense system
Given the complexity of the cell signaling which is triggered by the components of cigarette smoke, it is often difficult to isolate the effects of a specific component with regards to the pulmonary pathobiology of COPD. Therefore, most of the studies rely on the pathobiological effects of cigarette smoke rather than on its isolated components. In addition to the activation of several oxidant producing systems and enzymes in COPD, there is a simultaneous decline/inactivation of many antioxidant enzymes in the COPD lung [38], which further increases the oxidant burden. Ultimately, the increased oxidant burden causes an anoxidant/antioxidant imbalance, which is thought to play an important role in the pathogenesis of COPD.
THE PATHOBIOLOGY OF COPD
Extensive research which has been done, has highlighted the role of ROS/RNS in the pathobiology of COPD [39, 40]. The ROS/RNS induce alterations in the cellular macromolecules, which include lipid peroxidation, DNA damage and the inactivation of proteins (including enzymes) [39]. For instance, the ROS/RNS can oxidize a critical methionine residue in the active centre of α1-AT (Met358), thus forming methionine sulfoxide, with a loss of the biological activity of the enzyme. In addition to the above, the ‘nitrosative stress’ which is produced by the increased generation of RNS (e.g. nitric oxide, peroxynitrite etc.) in COPD may contribute to the inactivation of α1-AT by the nitration of the tyrosine residues in the protein molecule [40]. Other anti-proteases, e.g. tissue inhibitors of the matrix metalloproteinases, and enzymes such as lysyl oxidase (which are essential for the cross-linking of the matrix proteins, i.e. collagen and elastin) may be similarly damaged [41]. Furthermore, the aldehydes which are produced during lipid peroxidation are not innocent bystanders; they form adducts with the proteins, which include enzymes, and DNA [42]. Therefore, in chronic smokers, several mechanisms operate to reduce the biological activity of the anti-proteases, thereby compromising the local pulmonary antiprotease defense system. This tips the balance in favour of net proteolysis, matrix destruction and subsequent emphysema [43].
The enzymes such as elastase, cathepsins and matrix metalloproteinases (MMPs) can collectively degrade virtually all the matrix components, e.g. elastin, collagen, proteoglycans, laminin and fibronectin [37,41,43]. Neutrophil elastase has the added ability to: (i) increase the production of interleukin-8 (a neutrophil chemo attractant cytokine) by the epithelial cells and its interaction with α1-AT to generate a chemo tactic activity; (ii) degrade the lung immunoglobulins; (iii) damage the respiratory epithelium; (iv) stimulate the mucus gland hyperplasia; and (v) reduce the ciliary beating, thus impairing the host defense and favouring mucus retention [30,37,44]. The oxidative stress-mediated inflammatory response is also associated with a reduced neutrophil deformability [45], the up-regulation of various cell-adhesion molecules, an enhanced expression of the important transcription factors such as nuclear factor-kappa B (NF-κB, that in turn, leads to an increased expression of the pro-inflammatory mediators which include cytokines, e.g. tumour necrosis factor-α) [46] and apoptosis of the endothelial/epithelial cells. There is an extensive ‘cross-talk’ between the cells which participate in the inflammatory response (neutrophils, macrophages, lymphocytes and eosinophils) and the cells which form the structural components of the lungs (epithelial cells, endothelial cells, fibroblasts, etc. [47]. Both the T and B lymphocytes participate in the inflammatory response, and are together responsible for the persistence of the pulmonary inflammation, even in the absence of the primary insult [48]. To make things worse, the inactivation of α1-AT interferes with the normal functioning of the pulmonary surfactant [49]. The ‘triad’ of mucus gland hyperplasia, mucus retention and surfactant dysfunction produces dire consequences on the pulmonary gas-exchange. The inflammatory response has prompted some researchers to propose the ‘inflammatory/anti-inflammatory’ hypothesis for COPD, that also provides a sound basis for the use of anti-inflammatory drugs in the disease [50].
The lungs of the COPD patients are an easy prey for various micro-organisms, which leads to recurrent infections. This causes periodic exacerbations in COPD and each of such episodes perpetuate the lung damage by immunological as well as non-immunological mechanisms [51]. These exacerbations represent an imbalance in the lower airway bacterial colonization and the host defenses because of: (i) the acquisition of a new bacterial strain(s) which is not recognized by the host defenses; (ii) a change in the antigenic profile of the colonizing strain; and (iii) reduced host defenses due to viral infections or air pollution. Apart from the bacterial colonization and invasion, there is also a delayed bacterial clearance from the body, which is attributable to a lazy immune response, both cellular as well as humoral [48]. In addition to the above, the recurrent infections are likely to tip the oxidant/antioxidant balance in favour of the oxidants.
In order to cope with the oxidant challenge, evolution has endowed the cells with preventive, interceptive and repair mechanisms which are termed as the Antioxidant Defense System (ADS). The defense system can be categorized into protection via enzymatic activities and protection, through Low Molecular Weight Antioxidants (LMWA) [52]. However, the activities of various enzymes as well as the concentrations of LMWA which are involved in the detoxification of ROS/RNS have been reported to be low in COPD, thus indicating a suboptimal potential of the ADS [53]. Furthermore, the oxidant/antioxidant imbalance is also responsible for the corticosteroid resistance, as well as certain extrapulmonary manifestations in many COPD patients, e.g. atherosclerosis, muscular weakness and cachexia [54].
Oxygen is frequently administered as a therapeutic intervention during the acute exacerbations in COPD patients. Almost all tissues of the body can be injured by sufficiently high oxygen concentrations, but the lungs are exposed directly to the highest partial pressure of inspired oxygen [55]. The deleterious effects of hyperoxia are attributed to the various ROS which are the by-products of normal cellular oxidation-reduction processes, but their production increases markedly during hyperoxia. The high intracellular oxygen leads to the production of ROS from the pulmonary parenchymal cells and the phagocytes at a rate that overwhelms the natural cellular defenses. The overall result is death and lysis of the oxygen-sensitive cells, which result in microvascular and alveolar cell injuries which are typical of the oxygen toxicity. The rate at which the oxygen toxicity develops is directly related to the partial pressure of the inspired oxygen [56].
It is pertinent to ask- amidst the ongoing damage, why the lung is reluctant to repair itself. Normally, the adult lung is in a steady state where the amount of matrix which is produced is balanced by the amount which is degraded. Following a mild injury, the repair processes lead back to the steady state, however, if the insult is severe, the matrix follows either the ‘fibrotic program’ or the ‘erosion program’; the latter gets the upper hand in COPD [57]. Therefore, a heavy oxidant load in the pulmonary microenvironment is responsible not only for lung damage, but it also triggers a generalized erosion program, thus making the damage irreversible.
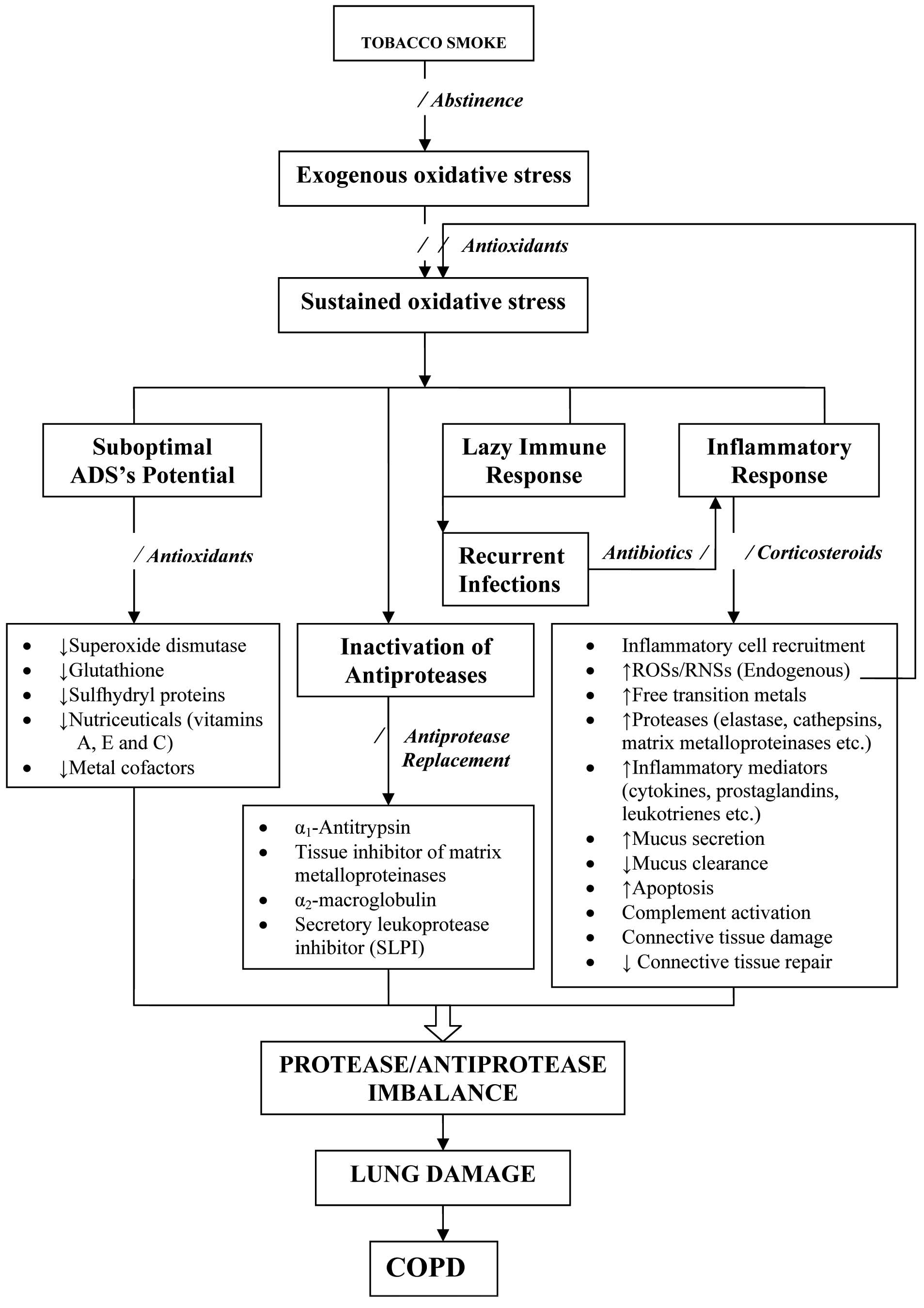
It is clear that the sustained oxidative/nitrosative stress in COPD patients is attributable to the increased exposure to exogenous oxidants (tobacco smoke), an increased generation of endogenous oxidants through the inflammatory response/recurrent infections/oxygen-therapy, a suboptimal and/or inappropriate antioxidant defense potential and last, but not the least, to the oxidants which generate more oxidants via chain reactions through the free transition metals and/or otherwise, as shown in [Table/Fig-3]. It is, therefore, reasonable to conclude that oxidative/nitrosative stress orchestrates the inflammatory response/abnormal immune functioning, which leads to lung damage, thus substantiating the oxidant/antioxidant hypothesis in COPD. Moreover, this hypothesis provides a rational basis to explain the pathogenic mechanisms in COPD and the plausible approach for therapeutic interventions [Table/Fig-5].
Schematic Representation of the Sequence of Events Underlying Pathobiology of COPD.
ADS: Antioxidant defense system
INTERVENTION STRATEGIES
Abstinence from smoking, the sooner the better, is worthwhile, because it consistently results in a reduction in the cough, expectoration, and the acute respiratory infections, but the patients should not expect any improvement in the breathlessness [58]. Smoking cessation and long-term oxygen therapy are the only interventions which are known to alter the disease progression and improve the prognosis in some patients with COPD. The other indirect gains from abstinence, apart from the financial ones, are a reduction in the passive-smoking-induced lung diseases in adults and the respiratory problems in children, particularly asthma [59]. In young smokers with minor airways obstruction, there may be an improvement in the lung function on quitting smoking, but in middle age, the major effect is to slow the subsequent decline in the lung function, nearly to that of healthy, never-smokers. However, in advanced COPD, it is less certain that a further decline in the lung function is slowed, so that there is a strong case for an early therapeutic intervention.
Many drugs are known to be potentially useful in the treatment of COPD, but relatively few become available for the human use, due to a lack of safety and/or efficacy, or both [60]. The patients with COPD are conventionally treated with inhalers of short-acting bronchodilators during the mild stages of the disease or acute exacerbations. Mucolytic drugs, along with antibiotics, are useful for reducing the number and the length of the bronchopulmonary infections. Long-acting bronchodilators and inhaled corticosteroids are used when the COPD is stable, but the patient exhibits moderate to severe airflow limitation. Inhaled steroids, in combination with long-acting β2-adrenoceptor agonists, seem to reduce the number of exacerbations in the COPD patients and the rate of decline of the lung function [60, 61].
FUTURE PERSPECTIVES
The current therapies for COPD are far from being satisfactory, since they are unable to deter the downward trend in the pulmonary function. Many new compounds are being discovered, and a subset of these is being evaluated in humans. Until major findings occur, to further our understanding of the pathophysiology of COPD and surrogate markers [61] become available, our hopes reside on the new long acting bronchodilators and anti-inflammatory drugs (either inhaled or oral), leukotriene modifiers, antioxidants and a number of compounds which are aimed at treating different aspects of COPD, such as pulmonary hypertension and hypophosphataemia. In the near future, phosphodiesterase-4 inhibitors and recombinant antiproteases could be added to this armamentarium [35, 60].
The last three decades have witnessed the successful clinical use of antioxidants to augment the treatment plan of diverse conditions, which include inflammatory lung diseases. For example, the recombinant human superoxide dismutase (scavenger of O2.- radicals) has been shown to decrease the oxidative stress in ischaemia/reperfusion injuries [62]. The limitations in the clinical use of superoxide dismutase include its low tissue affinity and low stability in plasma. However, these are likely to be overcome by employing the lecithinized enzyme, as has been documented by its efficacy in pre-clinical studies which have been done on COPD [63]. Further, antioxidant therapy has been found to be a complete cure for chronic pancreatitis [64]. The ‘big three’ LMWA (β-carotene, ascorbate, and α-tocopherol) have similarly been shown to offer some protection against oxidative stress in COPD patients [65]. The same holds true for the COPD patients who require oxygen-therapy to counter the ‘hypoxia/hyperoxia’ injury which is attributed to ROSs/RNSs. Other compounds include N-acetyl cysteine (NAC) and ambroxol, precursors that help in replenishing the glutathione pool (GSH; a major intracellular non-enzymatic LMWA) [51,66]. Interestingly, to maintain the cellular thiol pool, recombinant thioredoxin is emerging as a therapeutic option in oxidative stress-mediated lung diseases [67]. Antioxidants are known to prevent the oxidative damage besides down-regulating the inflammatory response in COPD [68]. It is reasonable to suggest that antioxidants (oxidant-antidotes) may become a universal part of the physicians’ prescription, together with the standard therapeutic regimen, not only in COPD, but also in other clinical conditions where an element of oxidative stress is present.
[1]. Celli BR, Mac Nee W, Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paperEur Respir J 2004 23:932-46. [Google Scholar]
[2]. American Thoracic SocietyEuropean Respiratory Society ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxideAm J Respir Crit Care Med 2005 171:912-30. [Google Scholar]
[3]. Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS, Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summaryAm J Respir Crit Care Med 2001 163:1256-76. [Google Scholar]
[4]. Wright JL, Emphysema: concepts under change – a pathologist’s perspectiveMod Patho 1995 18:873-80. [Google Scholar]
[5]. Fletcher C, Peto R, The natural history of chronic airflow obstructionBr Med J 1997 1:1645-48. [Google Scholar]
[6]. Kim WD, Eidelman DH, Izquierdo JL, Ghezzo H, Saetta MP, Cosio MG, Centrilobular and panlobular emphysema in smokers. Two distinct morphologic and functional entitiesAm Rev Respir Dis 1991 144:1385-90. [Google Scholar]
[7]. Soler-Cataluna JJ, Martinez-Garcia MA, Roman Sanchez P, Salcedo E, Navarro M, Ochando R, Severe acute exacerbations and mortality in patients with chronic obstructive pulmonary diseaseThorax 2005 60:925-31. [Google Scholar]
[8]. Anthonisen NR, Manfreda J, Warren CP, Hershfield ES, Harding GK, Nelson NA, Antibiotic therapy in exacerbations of chronic obstructive pulmonary diseaseAnn Intern Med 1987 106:196-204. [Google Scholar]
[9]. Calverley PM, Walker P, Chronic obstructive pulmonary diseaseLancet 2003 362:1053-61.GOLD (2010) Global Initiative for Chronic Obstructive Lung Disease (GOLD) [Google Scholar]
[10]. Halbert RJ, Isonaka S, George D, Iqbal A, Interpreting COPD prevalence estimates: what is the true burden of disease?Chest 2003 123:1684-92. [Google Scholar]
[11]. Menezes AM, Perez-Padilla R, Jardim JR, Muino A, Lopez MV, Valdivia G, Montes de Oca M, Talamo C, Hallal PC, Victora CG, Chronic obstructive pulmonary disease in five Latin American cities (the PLATINO study): a prevalence studyLancet 2005 366:1875-81. [Google Scholar]
[12]. Bano R, Ahmad N, Mahagaonkar AM, Latti RG, Study of lung functions in smokers and non-smokers in rural IndiaIndian J Physiol Pharmacol 2011 55:84-88. [Google Scholar]
[13]. Maitra A, Kumar V, The lung and the upper respiratory tract. In: Kumar V, Cotran RS, Robbins SL, EdsRobbins Basic Pathology 2003 7thNew DelhiSaunders (Reed Elsevier India Pvt. Ltd.):453-508. [Google Scholar]
[14]. Jindal SK, Aggarwal AN, Chaudhry K, Chhabra SK, D’Souza GA, Gupta D, A multicentric study on epidemiology of chronic obstructive pulmonary disease and its relationship with tobacco smoking and environmental tobacco smoke exposureIndian J Chest Dis Allied Sci 2006 48:23-29. [Google Scholar]
[15]. Peto R, Smoking and death: The past 40 years and the next 40BMJ 1994 309:937-40. [Google Scholar]
[16]. Snider G, Chronic obstructive pulmonary disease: risk factors, pathophysiology and pathogenesisAnnu Rev Med 1989 40:411-29. [Google Scholar]
[17]. Lundback B, Lindberg A, Lindstrom M, Ronmark E, Jonsson AC, Jonsson E, Larsson LG, Andersson S, Sandstrom T, Larsson K, Not 15 but 50% of smokers develop COPD? – Report from the Obstructive Lung Disease in Northern Sweden StudiesRespir Med 2003 97:115-22. [Google Scholar]
[18]. Kotaniemi JT, Pallasaho P, Sovijarvi AR, Laitinen LA, Lundback B, Respiratory symptoms and asthma in relation to cold climate, inhaled allergens, and irritants: a comparison between northern and southern FinlandJ Asthma 2003 39:649-58. [Google Scholar]
[19]. Koyama H, Geddes DM, Genes, oxidative stress, and the risk of chronic obstructive pulmonary diseaseThorax 1998 53(Suppl):10S-14S. [Google Scholar]
[20]. Johnson P, Balakrishnan K, Ramaswamy P, Ghosh S, Sadhasivam M, Abirami O, Prevalence of chronic obstructive pulmonary disease in rural women of Tamilnadu: implications for refining disease burden assessments attributable to household biomass combustionGlob Health Action 2011 4:26-72. [Google Scholar]
[21]. Marvisi M, Civardi G, Chronic obstructive pulmonary disease beyond cigarette smokeRespiration 2005 72:575-76. [Google Scholar]
[22]. Oberley-Deegan RE, Regan EA, Kinnula VL, Crapo JD, Extracellular superoxide dismutase and risk of COPDCOPD 2009 6:307-12. [Google Scholar]
[23]. Montes de Oca M, Halbert RJ, Lopez MV, Perez-Padilla R, Tálamo C, Moreno D, Chronic bronchitis phenotype in subjects with and without COPD: the PLATINO studyEurRespir J 2012 Jan 26 [Epub ahead of print, PMID:22282547] [Google Scholar]
[24]. Clarke PBS, Tobacco smoking, genes, and dopamineLancet 1998 352:84-85. [Google Scholar]
[25]. Lakhdar R, Denden S, Kassab A, Leban N, Knani J, Lefranc G, Update in chronic obstructive pulmonary disease: role of antioxidant and metabolizing gene polymorphismsExp Lung Res 2011 37:364-75. [Google Scholar]
[26]. Zhang RB, He QY, Yang RH, Lu BB, Liu YJ, Study on matrix metalloproteinase 1, 9, 12 polymorphisms and susceptibility to chronic obstructive pulmonary disease among Han nationality in northern ChinaZhonghua Liu Xing Bing Xue Za Zhi 2005 26:907-10. [Google Scholar]
[27]. Lee YL, Chen W, Tsai WK, Lee JC, Chiou HL, Shih CM, Polymorphisms of p53 and p21 genes in chronic obstructive pulmonary diseaseJ Lab Clin Med 2006 147:228-33. [Google Scholar]
[28]. Knoell DL, Ralston DR, Coulter KR, Wewers MD, α1-antitrypsin and protease complexation is induced by lipopolysaccharide, interleukin-1β, and tumor necrosis factor-α in monocytesAm J Respir Crit Care Med 1998 157:246-55. [Google Scholar]
[29]. Senior RM, Anthonisen NR, Chronic obstructive pulmonary disease (COPD)Am J Respir Crit Care Med 1998 157(Suppl):S139-47. [Google Scholar]
[30]. Hoidal JR, McCusker KT, Marshall BC, Rao NV, Oxidative damage and COPD. In: Cherniack NS, EdChronic Obstructive Pulmonary Disease 1991 1stPhiladelphiaWB Saunders:44-49. [Google Scholar]
[31]. Halliwell B, Gutteridge JMC, Oxidative stress ; in Free radicals in biology and medicine, Halliwell B and Gutteridge JMC eds 1999 3rdNew YorkOxford University Press, Oxford University Press:246-350. [Google Scholar]
[32]. Pandey R, Gupta S, Lal H, Mehta HC, Aggarwal SK, Hyperhomocysteinemia and cardiovascular disease: the nutritional perspectivesIndian J ClinBiochem 2000 15(Suppl):20-30. [Google Scholar]
[33]. Eiserich JP, van der Vliet A, Handelman GJ, Halliwell B, Cross CE, Dietary antioxidants and cigarette smoke-induced biomolecular damage: a complex interactionAm J Clin Nutr 1995 62(Suppl):1490S-1500S. [Google Scholar]
[34]. Rahman I, Mac Nee W, Oxidant/antioxidant imbalance in smokers and chronic obstructive pulmonary diseaseThorax 1996 51:348-50. [Google Scholar]
[35]. Owen CA, Proteinase and oxidants as targets in the treatment of chronic obstructive pulmonary diseaseProc Am Thorac Soc 2005 2:373-85. [Google Scholar]
[36]. Isik B, Isik RS, Ceylan A, Calik O, Trace elements and oxidative stress in chronic obstructive pulmonary diseaseSaudi Med J 2005 26:1882-85. [Google Scholar]
[37]. Ciencewicki J, Trivedi S, Kleeberger SR, Oxidants and the pathogenesis of lung diseasesJ Allergy Clin Immunol 2008 122:456-68. [Google Scholar]
[38]. Harju T, Kaarteenaho-Wiik R, Soini Y, Sormunen R, Kinnula VL, Diminished immune reactivity of gamma-glutamylcysteine synthetase in the airways of smokers’ lungAm J Respir Crit Care Med 2002 166:754-59. [Google Scholar]
[39]. Aoshiba K, Zhou F, Tsuji T, Nagai A, DNA damage as a molecular link in the pathogenesis of COPD in smokersEur Respir J 2012 Jan 20 [Epub ahead of print, PMID:22267761] [Google Scholar]
[40]. Sugiura H, Ichinose M, Nitrative stress in inflammatory lung diseasesNitric Oxide 2011 25:138-44. [Google Scholar]
[41]. Travis J, Oxidants and antioxidants in the lungAm Rev Respir Dis 1987 135:773-74. [Google Scholar]
[42]. Stoian I, Oros A, Moldoveanu E, Apoptosis and free radicalsBiochem Mol Med 1996 59:93-97. [Google Scholar]
[43]. Fischer BM, Pavlisko E, Voynow JA, Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammationInt J Chron Obstruct Pulmon Dis 2011 6:413-21. [Google Scholar]
[44]. Rahman I, Oxidative stress in pathogenesis of chronic obstructive pulmonary disease: cellular and molecular mechanismsCell Biochem Biophys 2005 43:167-88. [Google Scholar]
[45]. MacNee W, Selby C, New perspectives on basic mechanisms in lung disease 2- Neutrophil traffic in the lungs: role of hemodynamics, cell adhesion, and deformabilityThorax 1993 48:79-88. [Google Scholar]
[46]. Christman JW, Sadikot RT, Blackwell TS, The role of nuclear factor-κB in pulmonary diseasesChest 2000 117:1482-87. [Google Scholar]
[47]. Haslett C, Inflammation and the lung. In: Weatherall DJ, Ledingham JGC, Warrel DA, EdsOxford Textbook of Medicine 1996 3rdOxfordOxford University Press:2616-28. [Google Scholar]
[48]. Van der Strate BWA, Postma DS, Brandsma C-A, Melgert BN, Luinge MA, Geerlings M, Cigarette smoke-induced emphysema: A role for the B cell?Am J Respir Crit Care Med 2006 173:751-58. [Google Scholar]
[49]. Belai Y, Hernandez-Juviel JM, Bruni R, Waring AJ, Walther FJ, Addition of alpha1-antitrypsin to surfactant improves oxygenation in surfactant deficient ratsAm J Respir Crit Care Med 1999 159:917-23. [Google Scholar]
[50]. Papi A, Luppi F, Franco F, Fabbri LM, Pathophysiology of exacerbations of chronic obstructive pulmonary diseaseProc Am Thorac Soc 2006 3:245-51. [Google Scholar]
[51]. Han MK, Martinez FJ, Pharmacotherapeutic approaches to preventing acute exacerbations of chronic obstructive pulmonary diseaseProc Am Thorac Soc 2011 8:356-62. [Google Scholar]
[52]. Chevion S, Chevion M, Antioxidant status and human health: Use of cyclic voltammetry for the evaluation of the antioxidant capacity of plasma and of edible plants. In: Chiueh CC, EdReactive Oxygen Species: From Radiation to Molecular Biology 2000 New YorkNew York Academy of Sciences:308-25. [Google Scholar]
[53]. Nicks ME, O’Brien MM, Bowler RP, Plasma antioxidants are associated with impaired lung function and COPD exacerbations in smokersCOPD 2011 8:264-69. [Google Scholar]
[54]. Morley JN, Thomas DR, Wilson M-MG, Cachexia: pathophysiology and clinical relevanceAm J ClinNutr 2006 83:735-43. [Google Scholar]
[55]. Deneke SM, Fanburg BL, Normobaric oxygen toxicity of the lungN Engl J Med 1980 303:76-86. [Google Scholar]
[56]. Jackson RM, Pulmonary oxygen toxicityChest 1985 88:900-05. [Google Scholar]
[57]. Skold CM, Liu X, Umino T, Human neutrophil elastase augments fibroblast-mediated contraction of released collagen gelsAm J Respir Crit Care Med 1999 159:1138-46. [Google Scholar]
[58]. Pride NB, Stockley RA, Chronic obstructive pulmonary disease. In: Weatherall DJ, Ledingham JGC, Warrel DA, EdsOxford Textbook of Medicine 1996 3rdOxfordOxford University Press:2766-79. [Google Scholar]
[59]. Calverley PMA, Smoking cessation: professional problem or public policy?Respir Med 1996 90:123-25. [Google Scholar]
[60]. Molfino NA, Drugs in clinical development for chronic obstructive pulmonary diseaseRespiration 2005 72:105-12. [Google Scholar]
[61]. Houben JM, Mercken EM, Ketelslegers HB, Bast A, Wouters EF, Hageman GJ, Telomere shortening in chronic obstructive pulmonary diseaseRespir Med 2009 103:230-36. [Google Scholar]
[62]. Ambrosio G, Weisfeldt ML, Jacobus WE, Flaherty JT, Evidence for a reversible oxygen-radical-mediated component of reperfusion injury: reduction by recombinant human superoxide dismutase administered at the time of reflowCirculation 1987 75:282-91. [Google Scholar]
[63]. Tanaka K, Tanaka Y, Miyazaki Y, Namba T, Sato K, Aoshiba K, Therapeutic effect of lecithinized superoxide dismutase on pulmonary emphysemaJ Pharmacol Exp Ther 2011 338:810-18. [Google Scholar]
[64]. Whiteley G, Kienle A, Lee S, Taylor P, Schofield D, Braganza J, Micronutrient antioxidant therapy in the non-surgical management of chronic pancreatitis: long-term observationsPancreas 1994 9:A807 [Google Scholar]
[65]. Grievink L, Smit HA, Ocke MC, van’t Veer P, Kromhout D, Dietary intake of antioxidant (pro)-vitamins, respiratory symptoms and pulmonary function: the MORGEN studyThorax 1998 53:166-71. [Google Scholar]
[66]. Gillisen A, Nowak D, Characterization of N-acetylcysteine and ambroxol in anti-oxidant therapyRespir Med 1998 92:609-23. [Google Scholar]
[67]. Xu J, Li T, Wu H, Xu T, Role of thioredoxin in lung diseasePulm Pharmacol Ther 2012 Jan 25 [Epub ahead of print, PMID:22293327] [Google Scholar]
[68]. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim Biophys Acta 2011 Nov 9 [Epub ahead of print, PMID:22101076 [Google Scholar]