A Consanguinity Related Autosomal Translocation which Leads to Premature Ovarian Failure
Mot Yee Yik1, Murizah Mohd Zain2, Zubaidah Zakaria3, Narazah Mohd Yusoff4
1 PhD Student, Advanced Medical and Dental Institute, Universiti Sains Malaysia.
2 Fellow in Reproductive Medicine, Consultant Obstetrician and Gynaecologist, Reproductive Medicine Specialist, Infertility Unit, Hospital SultanahBahiyah.
3 Consultant Pathologist, Institute for Medical Research.
4 Consultant Haematologist, Associate Professor, Advanced Medical and Dental Institute, Universiti Sains Malaysia.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Narazah Mohd Yusoff, Regenerative Medicine Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, No. 84, PersiaranSeksyen 4/9, Bandar Putra Bertam, 13200 Kepala Batas, SeberangPerai Utara, Pulau Pinang, Malaysia.
E-mail: narazah@amdi.usm.edu.my
The premature ovarian failures with underlying chromosomal abnormalities are normally X-linked, although their associations with the autosomal and the Robertsonian translocations are also possible. Here, we are reporting a case of premature ovarian failure which was associated with a translocation between the long arm of chromosome 7 at q11.23 and the short arm of chromosome 5 at p15.3. The proband was a 26-year-old Malay woman who presented with premature ovarian failure, who was referred for cytogenetic testing due to the suspicion of a chromosomal anomaly. Her physical examination revealed that she had no abdominal or pelvic masses and that she had normal secondary sexual characteristics. Her medical history as well, revealed no points for concern. However, a consanguineous relationship existed, as the patient’s paternal grandmother and maternal grandfather were biological cousins. Our present case indicated that region p15.3 of chromosome 5 and region q11.23 of chromosome 7 possibly carried essential genes for the ovarian function and that they postulated a link between the consanguinity and the chromosomal abnormalities.
Premature ovarian failure, Chromosomal abnormality, Consanguinity, Translocation, Cytogenetic test
INTRODUCTION
Premature Ovarian Failure (POF) is a state of an impaired ovarian function in women, that can be principally divided into two groups: primary and secondary amenorrhoea. Primary amenorrhoea is characterized by the absence of menarche which is due to a primary ovarian defect, while secondary amenorrhoea is presented as a premature depletion of the ovarian follicles or an arrest in folliculogenisis under the age of 40 years. The incidence of POF is one in 10,000 women by the age of 20 years and it affects up to one in 100 women by the age of 40 years [1]. The causative factors of POF can be classified as chromosomic and non-chromosomic defects. The POFs which are associated with chromosomal abnormalities are normally X-linked [2,3], although reports of autosomal [4] and Robertsonian translocations [5] have also been published. The non-chromosomal abnormality factors can be of either an iatrogenic, autoimmune, infectious or an idiopathic origin. Consanguinity has been widely speculated as a significant contributing factor to the chromosomal abnormalities. The available literature suggests that the offspring of consanguineous unions may have increased chances of a genetic homozygosity and other anomalies which include chromosomal numerical and structural defects [6]. Such genetic aberrations may result in various phenotypic presentations, which include POF.
CASE REPORT
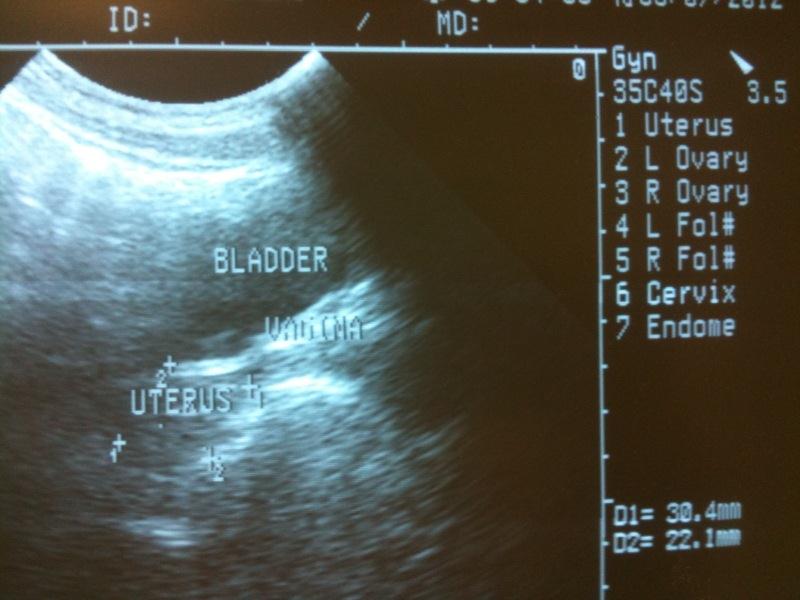
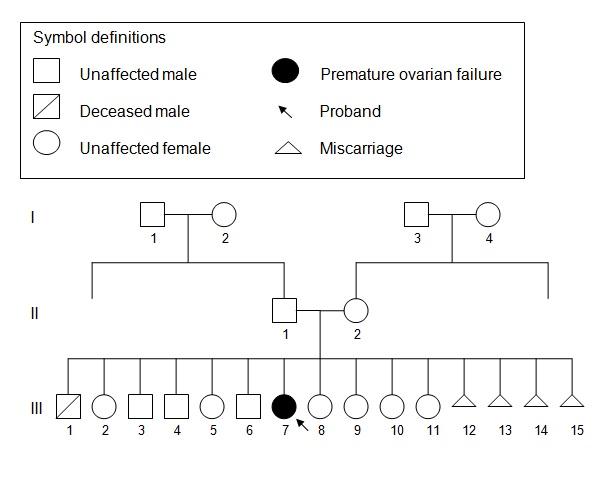
A 26-year-old Malay woman was referred for cytogenetic tests at the Advanced Diagnostic Laboratory (ADL) due to the suspicion of a chromosomal anomaly upon a preliminary diagnosis of Premature Ovarian Failure (POF) at the Hospital Sultanah Bahiyah’s Infertility Clinic. A hormonal assay which was done, revealed elevated levels of FSH (101.8 mU/mL) and LH (60.3 mU/mL). The TSH, T4 and the Prolactin levels were normal. A transabdominal ultrasound was performed on the patient, which revealed that she has a small uterus, as has been indicated in [Table/Fig-1]. Her physical examination revealed that she has no abdominal or pelvic masses and that her secondary sexual characteristics were also normal. Her medical history as well, revealed no points for concern. However, a consanguineous relationship existed, as the patient’s paternal grandmother and maternal grandfather were biological cousins. As has been shown in [Table/Fig-2], the family’s pedigree revealed that the proband (III.7) was of the first generation from the said consanguinity and that therefore, any anomalies that would result, would be manifested in her sibship. Her mother (II.2) had 15 pregnancies which had resulted in four boys, seven girls and four late miscarriages.
Transabdominal ultrasound image of the patient reveals the presence of a small uterus
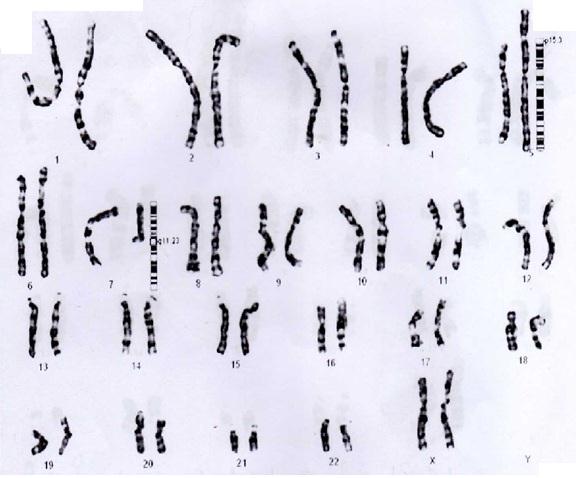
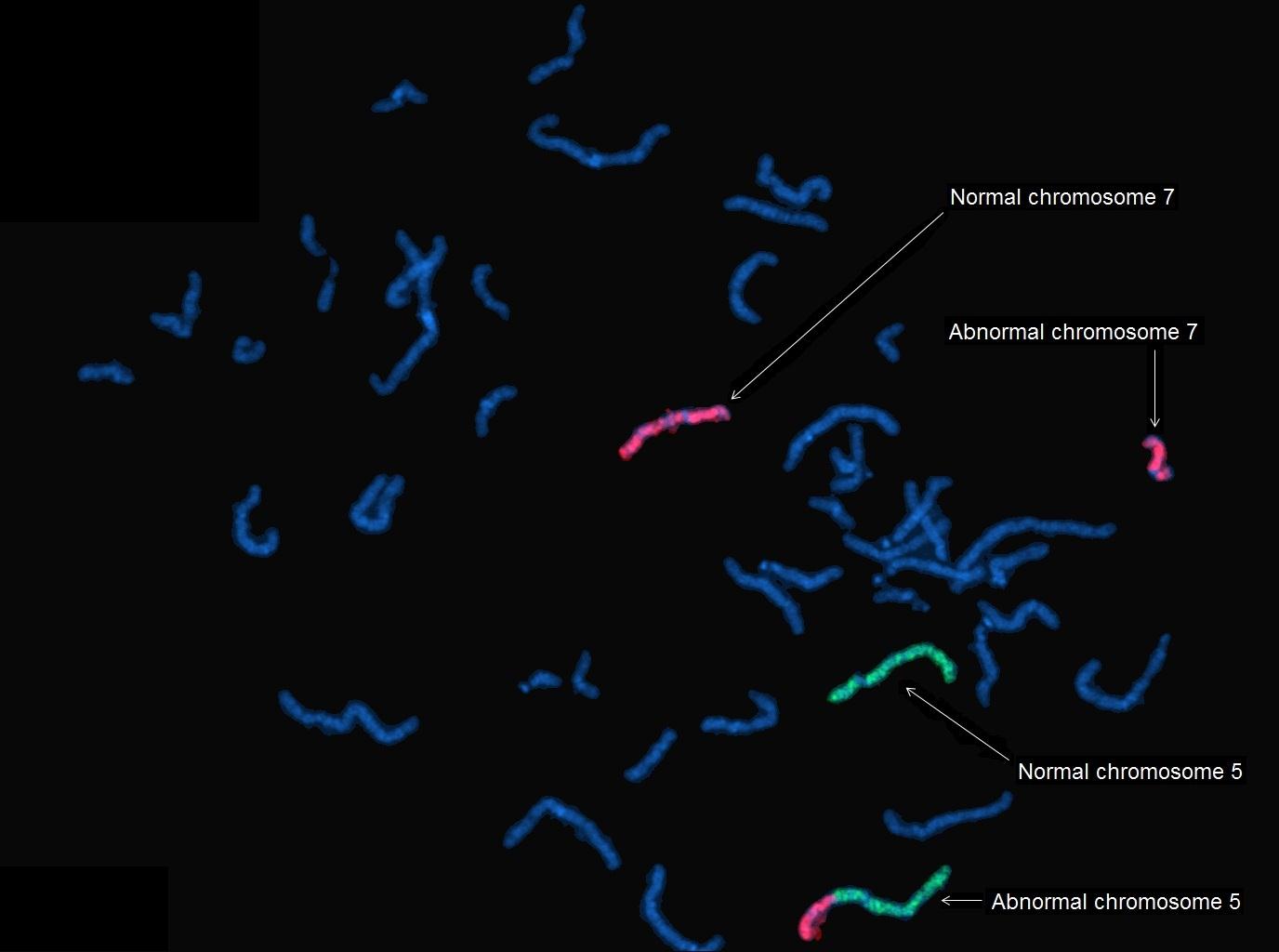
A cytogenetics test was requested on peripheral blood leukocytes from the proband. A standard procedure for the chromosomal analysis was performed on the cultured cells by using Giemsa banding (G-banding). Fluorescence In Situ Hybridization (FISH) was performed for the confirmation of the abnormality. Karyotype analyses from 18 G-banded metaphase cells were performed for this case. The proband exhibited a balanced translocation between chromosome 5 and 7, with a karyotype of 46, XX, t(5;7)(q11.23;p15.3), as was indicated by the cytogenetic analysis and 356this was confirmed by Whole Chromosome Paint (WCP). The complete karyotype and the breakpoints of the chromosomes are illustrated in [Table/Fig-3]. The FISH image with the WCP of chromosomes 5 (spectrum green) and 7 (spectrum orange) are shown in [Table/Fig-4].
Karyotype of the proband illustrating a single autosomal translocation between chromosome 5 (p15.3) and chromosome 7 (q11.23).
[Table/Fig-4] illustrates that the abnormal chromosome 5 with the translocated material from chromosome 7 is indicated as a bicoloured chromosome. The abnormal chromosome 7 is observed as a truncated form of the normal chromosome 7 as the translocated material from chromosome 5 is very close to the end of the p arm and it may not be clearly visualized.
FISH image with whole chromosome paint (WCP) of chromosome 5 (spectrum green) and chromosome 7 (spectrum orange).
DISCUSSION
The chromosomal abnormalities which result in POF have been most commonly associated with the X-chromosome due to the localization of essential ovarian function genes in the regions of POF1 at Xq26-q27 [7] and POF2 at Xq13.3-q21.1 [8]. The increasingly autosomal abnormalities [3] which include Robertsonian translocations [4] have been implicated to the POF incidences. These findings suggest that the critical genes which are involved in the ovarian function may be found on the autosomal loci as well.
The present case demonstrated a translocation between the long arm of chromosome 7 with a breakpoint at q11.23 and the short arm of chromosome 5 at p15.3. The translocation which has been described here was most likely, a non-inherited, spontaneous event with the possibility of consanguinity as the causal factor. The details which supported these notions included the absence of previous reports of POF in the family, a large sibship, a single translocation and the proband being the first generation offspring of the consanguineous marriage. Other than the presentation of POF, the proband has no other physical anomalies. It was plausible that the breakpoint(s) of this translocation encoded an essential ovarian function gene(s), which had resulted in POF.
Our present case was in agreement with the reports of the current literature, that the POF related genes were found on both chromosome 5 [9] and chromosome 7 [10]. The study of Oldenburg et al., [9] revealed that a study which was done on a Dutch family had identified a POF susceptibility region on chromosome 5, at q14.1-q15. Caburet et al., [10] determined that the region of chromosome 7q21-22 harboured a locus which was common in the familial cases of POF. A recent report which was made by Caburet et al., [11] has revealed two novel loci on chromosome 7 in a highly consanguineous pedigree, whereby the genes which were associated with the presentation of POF could reside. The loci which were identified were on chromosome 7p21.1-15.3 and 7q21.3-22.2. Although the translocation in our case was of the mentioned chromosomes, the loci which were identified were different. It would be interesting to further investigate the translocation breakpoints to determine the disrupted genes and to identify their correlation to the ovarian function. On a wider perspective, additional studies will be required to determine whether the chromosomes which have been discussed in this report could play more essential roles in POF than it was previously thought.
Consanguinity has been reportedly linked to increased risks of mental retardation and multiple congenital malformations [12]. A recent study which was done by Amudha et al., [6] provided significant data to suggest the role of consanguinity in the occurrence of chromosomal abnormalities, which included numerical and structural defects. Our present case proposed that consanguinity may lead to translocations which are capable of manifesting POF, possibly via the disruption of essential genes on the chromosomal breakpoints. Amudha et al., [6] further postulated that there could be a relationship between the types of consanguinity and the chromosomal abnormalities that would arise in the offspring. The validation of such connections would be helpful in the genetic counselling which is aimed at preventing the occurrence of the chromosomal abnormalities which include those which are related to POF. Additional studies which are done on larger sample sizes and an improved research design in the communities where consanguinity is commonly practised, may provide invaluable insights to substantiate the role of consanguinity on the chromosomal abnormalities which are related to POF.
[1]. Coulam CB, Adamson SC, Annegers JF, Incidence of premature ovarian failureObstet Gynecol. 1986 67:604-06. [Google Scholar]
[2]. Artini PG, Ruggiero M, Papini F, Valentino V, Uccelli A, Cela V, Chromosomal abnormalities in women with premature ovarian failureGynecological Endocrinology 2010 26(Suppl 10):717-24. [Google Scholar]
[3]. Goswami D, Conway GS, Premature Ovarian FailureHum Reprod. Update 2005 11:391-410. [Google Scholar]
[4]. Burton KA, Van Ee CC, Purcell K, Winship I, Shelling A, Autosomal translocation associated with premature ovarian failureJ Med Genet. 2000 37(Suppl 5):e2 [Google Scholar]
[5]. Kawano Y, Narahara H, Matsui N, Miyakawa I, Premature ovarian failure associated with a Robertsonian translocationActa Obstet Gynecol Scand. 1998 77:467-69. [Google Scholar]
[6]. Amudha S, Aruna N, Rajangam S, Consanguinity and chromosomal abnormalityIndian Journal of Medical Genetics 2005 11(Suppl 2):108-10. [Google Scholar]
[7]. Tharapel AT, Anderson KP, Simpson JL, Martens PR, Wilroy RS Jr, Llerena JC Jr, Deletion (X)(q26.1-q28) in a proband and her mother: molecular characterization and phenotypic-karyotypic deductionsAm J Hum Genet 1993 52:463-71. [Google Scholar]
[8]. Powell CM, Taggart RT, Drumheller TC, Wangsa D, Qian C, Nelson LM, Molecular and cytogenetic studies of an X;autosome translocation in a patient with premature ovarian failure and review of the literatureAm J Med Genet. 1994 52:19-26. [Google Scholar]
[9]. Oldenburg RA, van Dooren MF, de Graaf B, Simons E, Govaerts L, Swagemakers S, A genome-wide linkage scan in a Dutch family identifies a premature ovarian failure susceptibility locusHuman Reproduction 2008 23(Suppl 12):2835-41. [Google Scholar]
[10]. Caburet S, Laissue P, Christin-Maitre S, Bouchard P, BenNeriah Z, Shavel S, Identification of a novel locus on chromosome 7 in familial cases of premature ovarian failureASHG Annual Meeting 2006 :2175 [Google Scholar]
[11]. Caburet S, Zavadakova P, Ben-Neriah Z, Bouhali K, Dipietromaria A, Charon C, Genome-wide linkage in a highly consanguineous pedigree reveals two novel loci on chromosome 7 for non-syndromic familial premature ovarian failurePLoS One 2012 7(3):e33412 [Google Scholar]
[12]. Kulkami ML, Kurian M, Consanguinity and its effect on fetal growth and development: a south Indian studyJ Med Genet 1990 27:348-52. [Google Scholar]