Fetal Haemoglobin and β-globin Gene Cluster Haplotypes among Sickle Cell Patients in Chhattisgarh
Sanjana Bhagat1, Pradeep Kumar Patra2, Amar Singh Thakur3
1 Assistant Professor, Departtment of Biochemistry and Biotechnology, Pt. J. N. M. Medical CollegeRaipur (C.G.), Chhattisgarh, India.
2 Professor and Head, Department of Biochemistry and Biotechnology, Pt. J. N. M. Medical CollegeRaipur (C.G.), Chhattisgarh, India.
3 Professor and Head, Department of Biochemistry, Govt Medical College, Jagdalpur (C.G.), India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Sanjana Bhagat, Department of Biochemistry and Biotechnology Pt. JawaharLal Nehru Memorial Medical College, Jail Road, Raipur (Chhattisgarh) 492001, India.
Phone: 09827118455
E-mail: sanjana_bhagat@rediffmail.com
Background: Foetal Haemoglobin (HbF) is the best-known genetic modulator of sickle cell anaemia, which varies dramatically in concentration in the blood of these patients. The patients with SCA display a remarkable variability in the disease severity. High HbF levels and the β-globin gene cluster haplotypes influence the clinical presentation of sickle cell disease. To identify the genetic modifiers which influence the disease severity, we conducted a β-globin haplotype analysis in the sickle cell disease patients of Chhattisgarh.
Aim: The foetal haemoglobin and the β-globin gene haplotypes of the sickle cell trait and the sickle cell disease patients from Chhattisgarh were investigated.
Materials and Method: A total of 100 sickle cell patients (SS), 50 sickle cell trait patients (AS) and 50 healthy control individuals were included in the present study. The distribution of the β-globin gene haplotype was done by the PCR-RFLP method.
Result: PCR-RFLP showed that the homozygous Arab-Indian haplotype (65%) was the most frequent one, followed by the heterozygous Arab-Indian haplotype (11%) in the sickle cell patients (SS), while the AS patients had a higher frequency of the heterozygous Arab-Indian haplotype (38%) in comparison to homozygous one (32%). Four atypical haplotypes, 3 Benin and 1 Cameroon were also observed, although they were in lower frequencies. In the present study, the HbF levels were higher in the AS and the SS patients, with one or two Arab-Indian haplotypes as compared to the other haplotypes.
Conclusion: The presence of the Arab-Indian haplotype as the predominant haplotype might be suggestive of a gene flow to/from Saudi-Arabia or India and it was associated with higher HbF levels and a milder disease severity.
Sickle cell anaemia, β-globin gene cluster haplotype, Foetal Haemoglobin
INTRODUCTION
Sickle cell disease, which is also known as Haemoglobin SS (HbS) disease, is caused by a mutation in the β-globin gene cluster. This mutation results in the production of an abnormal version of the beta chain of haemoglobin, which has difficulty in carrying oxygen properly through the body. However, this disease has been associated with a great phenotypic heterogeneity and clinical variability [1].
The HbS β-globin gene is found on four or five common haplotypes which reflect its regions of origin in different parts of Africa, the Middle East and the Indian subcontinent [2–4]. The β-globin gene cluster haplotype is a cis-acting determinant, which serves as a marker for the genetic background of the HbS disease patients and for predicting the disease severity. The Bantu haplotype has been correlated with the lowest level of HbF and those with a Senegal or a Saudi Arab-Indian haplotype have the highest levels of HbF [5–7]. The Cameroon and the Benin haplotypes have intermediate levels of HbF [6]. In this context, much work has been done for the analysis of the genetic determinants in the β gene cluster region that might affect the globin gene expression and thus relate to the clinical diversity of sickle cell disease. HbF is the most powerful modulator of the clinical and the haematologic features of Sickle Cell Anaemia (SCA) [8]. To protect against various complications of this disease, different concentrations of HbF were postulated to be required, although any increment in the HbF had a beneficial effect on the mortality. The Senegal and the Arab-Indian haplotypes are linked to the C-T polymorphism at position -158 in the Gγ promoter, that results in an increased number of Gγ chains and elevated HbF levels [9,10]. In addition, the Benin and the Bantu haplotypes lack this Single Nucleotide Polymorphism (SNP) [9,11].
Although the DNA haplotype in the β-globin gene can help in determining the genetic diversity of the specific population, the origin and the track of the gene flow [12]. Chhattisgarh, having a high density of tribes, came into existence as the 26th State of the Indian Union on first Nov. 2000. The population of Chhattisgarh is notable for the high proportion of scheduled tribes (31.76%). Furthermore, the previous data shows that the Chhattisgarh and Orissa states have the highest frequency of the HbS disease [13]. The presence of HbS was first reported by Lehmann and Cutbush [14] in the tribal populations of the Nilgiris, in the southern state of Tamil Nadu. Since then, a high incidence of HbS has also been reported in about 50 tribal populations in other areas of India [15,16]. To the best of our knowledge, no previous study has done to establish the HbS β-haplotype in the sickle cell disease and the sickle cell trait patients of Chhattisgarh.
In the present study, we have examined the β-globin haplotypes and their association with the HbF levels among the sickle cell trait and the sickle cell patients in Chhattisgarh.
MATERIALS AND METHODS
Hundred, non-consanguineous, sickle cell patients (SS) (51 males and 49 females; mean age 23.84 years ) and fifty sickle cell trait (AS) (25 males and 25 females; mean age 26.3 years ) were included in the study. They were from different regions (Raipur, Bilaspur, Durg, and Dhamtari) of Chhattisgarh, India. None of the patients were on any kind of treatment (other than folic acid supplementation), nor had any patient received blood transfusions during the 4 months, prior to the sample collection. An informed consent was obtained from all the subjects prior to the drawing of their blood samples. The blood samples from Chhattisgarh’s adult’s sickle cell trait and sickle cell disease patients were obtained by randomly sampling from a population survey and from some patients who had been admitted in the Dr. B. R. A. M. Hospital, Raipur. The control group was composed of 50 subjects (25 males and 25 females; mean age 24.88 years ) without sickle cell disease, who were living in different regions of Chhattisgarh and the DNA samples were collected randomly sampling by doing a population survey. The genomic DNA was then extracted by using the HiPuraA kit (Himedia).
The HbF levels were determined by using HPLC (VARIANTTM β – thalassemia Short Program (Bio-Rad). The haematological indices analysis was done by using a BC-3000Plus Auto Hematology Analyzer.
Seven well-established RFLP sites which included the HincII site of the 5’ε-globin (5’ε-HincII), the HindII sites at the intervening sequence II of the Gγ-globin (Gγ-IVSII-HindIII) and the Aγ-globin (Aγ-IVSII-HindIII), the HincII sites in ψβ-globin (ψβ-HincII), and the HincII site 3’of ψβ-globin (3’ψβ-HincII), the AvaII site in β-globin (β-AvaII) and the HinfI site in 3’β-globin (3’β-HinfI) were used to define the β-haplotype. The genomic DNA was first amplified by Polymerase Chain Reaction (PCR) by using published primers [17–19], followed by restriction digestion and agarose gel inspection. The presence of an A or T nucleotide at in the 6th codon of the β-globin was confirmed for all the DNA samples by using restriction enzyme DdeI analysis.
The RFLP data were used to construct the β-haplotype, based on the target gene specific digestion by the restriction enzyme and it was scored as +/+ if the two alleles were digested, as +/- if one but not the other allele was digested (heterozygote), and as -/- if no digestion occurred in the sample. The allele frequency was calculated by a direct gene counting method.
RESULT
The results of the haematological parameters which include total Hb, HbF, HCT, MCV and MCH in both the sexes (combined) for the AS and the SS patients are shown in [Table/Fig-1].
Haematological indices in both sexes among sickle cell trait and sickle cell disease patients.
| Haematological Parameter | Sickle Cell Disease (SS) n=100 | Sickle Cell Trait (AS) n=50 | Controls (AA) n=50 |
|---|
| Age in Years | 23.84 ±8.38 | 26.3 ±7.37 | 24.88 ±8.70 |
| HbF (%) | 19.39 ±7.14 | 1.06 ±0.74 | 0.12 ±0.13 |
| Hb (g/dl) | 9.29 ±2.54 | 12.01±2.56 | 12.59 ± 2.34 |
| HCT (%) | 29.53 ±10.40 | 32.08 ±5.55 | 34.86 ±6.54 |
| MCV(fl) | 91.29 ±10.88 | 77.88 ±10.56 | 81.41 ±10.03 |
| MCH(pg) | 29.71 ±8.67 | 28.32 ±5.87 | 28.11 ±4.32 |
Values are mean±SD; n = number of subjects.
[Table/Fig-2] presents the HbF levels with the haplotype distribution among the sickle cell disease and the sickle cell trait patients of Chhattisgarh, India. The Arab-Indian haplotype (+ + + - + + - +) was predominant in the Chhattisgarh population. Two major β-haplotypes were constructed for the SS patients, which included 65% of the homozygous Arab-Indian (+ + + - + + - +) and11% heterozygous Arab-Indian haplotypes, followed by the Arab- Indian in combination with the atypical haplotype, 5% Arab-Indian/ Cameroon (+ + + - + + - + / - - + + - + - +), 3% Arab-Indian/Benin (+ + + - + + - + /- - - - - + - +), 3% Arab-Indian/Bantu (+ + + - + + - + / - - + - - - - +) and 4% Cameroon/ Benin /(- - + + - + - + /- - - - - + - +) haplotype. Two rare haplotypes, 3 Benin and 1 Cameroon were also found in the study.
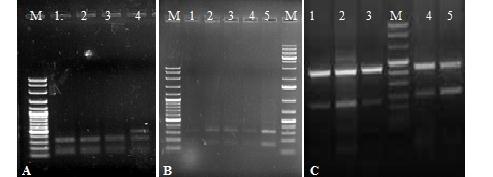
An agarose gel (A, B, C) shows electrophoresis pattern of RFLP product of β-Hinfl and ε-HincII site. Lane (M) represents of all three gel shows the molecular weight marker with 100 bp DNA ladder. In (Fig A) lane 1-3 and (Fig B) lane 1-5 shows RFLP product of (320 bp and 154bp ) with β-HinfI (-/-) site except in (Fig A) in which the lane 4 shows the HinfI (+/+) site with 213+107+154bp. In (Fig. C) lane 1, 2, 3, 4, 5 shows (+/+) sites for ε-HincII site.
Among 50 sickle cell trait (AS) patients who were studied, 32% were homozygous and 38% were heterozygous for the Arab-Indian haplotype. The atypical haplotype included 14% Arab-Indian/Benin (+ + + - + + - + /- - - - - + - +) and 10% Arab-Indian/Cameroon (+ + + - + + - + / - - + + - + - +). The remaining 6% of the AS patients inherited rare or incomplete haplotypes. The RFLP products of the different polymorphic sites have been shown in [Table/Fig-2 and 3].
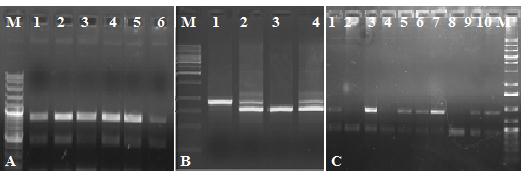
An agarose gel (A, B, C) shows electrophoresis pattern of RFLP product. In (Fig A) lane1-6 shows 3’ψβ-HindII site (+/-) site. In (Fig B) lane 1and lane 3shows absence (-/-) of Gγ-HindIII site, and lane 2 and lane 4 show presence of 3’ψβ-HindII site (+/+). In (Fig C) lane 1, 3, 5, 6, 7, 9 and10 shows presence of 5’3’ψβ-HindII site. Lane 2, 4 and 8 shows absence of β-AvaII site.
Among all the AS and the SS patients, the homozygous and the heterozygous Arab-Indian haplotypes were associated with the highest levels of HbF [Table/Fig-4].
Distribution of β-globin gene haplotypes and Fetal haemoglobin level amonsg sickle cell trait and sickle cell disease patients.
| Haplotype | SS (n=100) | HbF | AS (n=50) | HbF |
|---|
| Arab-Indian/Arab-Indian (+ + + - + + - + / + + + - + + +) | 65(65%) | 22.71±5.23 | 16 (32%) | 0.20±0.16 |
| Arab-Indian (Heterozygous) (+ + + - + + - +) | 11(11%) | 20.85±3.02 | 19 (38%) | 0.06±0.09 |
| Arab-Indian/Benin (+ + + - + + - + /- - - - - + - +) | 3 (3%) | 9.73±0.59 | 7 (14%) | 0.15±0.09 |
| Arab-Indian/Bantu (+ + + - + + - + / - - + - - - - +) | 3 (3%) | 3.83±0.46 | - | - |
| Arab-Indian/Cameroon (+ + + - + + - + / - - + + - + - +) | 5 (5%) | 12.84±0.67 | 5 (10%) | 0.06±0.08 |
| Benin/Benin (- - - - - + - + /- - - - - + - +) | 3 (3%) | 7.0±0.21 | - | - |
| Cameroon/Benin /(- - + + - + - + /- - - - - + - +) | 4 (4%) | 9.17±0.38 | - | - |
| Cameroon/Cameroon (- - + + - + - +/(- - + + - + - +) | 1(1%) | 10.1 | - | - |
| Rare 1 | 3 (3%) | 13.7±1.48 | 2 (4%) | 0.0 |
| Rare 2 | 2 (2%) | 14.7±0.1 | 1 (2%) | 0.0 |
SS, sickle cell disease; AS, sickle cell trait; HbF, fetal haemoglobin; n= number of subjects. HbF levels values are expressed in mean±SD.
DISCUSSION
This is the first report of the association of the HbF levels with the beta globin gene haplotypes in this region of India. In the present study, the Arab-Indian haplotype was found to be the most prevalent one among the SS patients, who included those with a homozygous form of the Arab-Indian haplotype (65%) and those with a heterozygous form (11%), while among the sickle cell trait (AS) patients, the heterozygous Arab- Indian (38%) haplotype was found in a higher frequency than the homozygous 1 (32%). This was consistent with the findings from earlier studies which were done in Andhra Pradesh and other parts of India [20–22]. However, four other atypical haplotypes, Benin, Bantu, Cameroon which were associated with the Arab-Indian and the Cameroon/ Benin haplotypes were also found in the present study. It was well documented that the presence of the atypical and the rare haplotypes such as Cameroon, Senegal and Benin was seen in the central and the southern parts of India [20,21]. We also found a rare Cameroon (1%) and a Benin (3%) haplotype in the SS patients of Chhattisgarh. The presence of these haplotypes was previously reported in India [20,23]. The predominant presence of the Arab- Indian haplotype which was linked to the βS gene in Chhattisgarh can be easily explained by the gene migration to/from Saudi Arabia and India with a genetic admixture. A recombination event or a gene conversion or a chance mutation at the RFLP loci may be the plausible explanations for the rare haplotypes which were found in our study. Also, the presence of the homozygous Benin and Cameroon haplotypes might be regarded as a reflection of the gene flow from Africa through the possible routes.
Despite the fact that SCA is caused by a single A-T mutation in the sixth codon of the β globin gene, there are many different clinical phenotypes which are associated with this monogenic disorder. The patients with elevated foetal haemoglobin levels generally have less severe disease. In our previous study, we found the presence of an XmnI (+/+) site in the SS and the AS patients, which was associated with an increase in the HbF (P<0.0001) synthesis [24]. In addition, we also found that the presence of one XmnI (+/-) site in the SS patients as compared to the XmnI-/- site, did not show any difference in the HbF levels. It is believed that the other genetic markers in the β-locus also exert a significant effect on the foetal haemoglobin synthesis [25]. In the carriers of the Senegal and the Arab-Indian haplotypes, the presence of an XmnI C-T polymorphism at the -158 position upstream of the Gγ gene was strongly associated with higher HbF levels [9,10]. In the present study, we found that most of the patients with the homozygous and the heterozygous Arab-Indian haplotypes had higher levels of HbF than those with other haplotypes. This finding was consistent with other reported influences of the Arab-Indian haplotype on the HbF level [7]. However, in our study, the presence of only chromosomes with the Arab-Indian haplotype was associated with high HbF levels in comparison to a combination with other haplotypes. These observations suggest that other factors which are perhaps linked to these haplotypes, are differentially affecting the gamma gene transcription.
In addition, we examined the effects of the five common haplotypes on the HbF variation in sickle cell (SS) and sickle cell trait patients in Chhattisgarh. Among the sickle cell patients, those who were homozygous for the Arab-Indian haplotype had high HbF values (22.71±5.23) as compared to the values (20.85±3.02) in the individuals who were heterozygous for the Arab-Indian haplotype. In the sickle cell trait (AS) patients, the patients with the homozygous Arab-Indian haplotype showed a high HbF level (0.20±0.16) as compared to those with the heterozygous haplotype (0.06±0.09). We confirmed the variation in the HbF levels which was associated with the different haplotypes which we found in the present study. The beta globin gene haplotypes were used as a markers for the phenotypic heterogeneity of sickle cell disease, because of their association with the variable HbF levels. However, the influences of the HBB locus and the XmnI-HBG2 site on the HbF levels in SCA have been validated by many studies in several populations.
The SCA patients with the Benin haplotype often have low HbF levels, while those with the Cameroon haplotype show intermeditary HbF levels. However, our results were in agreement with some previously published data on the HbF levels and the βS-globin haplotype. In our study, the presence of the intermediate HbF levels for the CAR/CAR haplotype could be due to the sequence variations in the regulatory regions, such as the 5’HS2 and the flanking region of the γ gene [26]. Our results showed that the differences in the HbF levels among the SCA patients were associated with the Cameroon/ Benin haplotype (HbF = 9.17±0.38), while the Cameroon/ Cameroon haplotype (HbF =10.1), and the Cameroon/ Arab-Indian haplotype (HbF = 12.84±0.67), exhibited higher HbF levels and a milder disease presentation. In our study, we found significant differences in the HbF levels between the Cameroon and the Benin haplotypes.
CONCLUSIONS
Our study confirmed the high diversity of the βS-globin gene haplotypes and the high level of phenotypic heterogeneity among the sickle cell anaemia patients in Chhattisgarh, India. In conclusion, this study determined the unicentric origin of the sickle mutation in this region, in which the Arab- Indian haplotype exists in excellent agreement with the historical record. In addition, we demonstrated that the atypical haplotypes which were observed in association with the sickle cell gene were likely to be generated by diverse genetic mechanisms which involved either the Cameroon or the Benin and the Bantu haplotypes.
*Values are mean±SD; n = number of subjects.
*SS, sickle cell disease; AS, sickle cell trait; HbF, fetal haemoglobin; n= number of subjects. HbF levels values are expressed in mean±SD.
[1]. Steinberg MH, Predicting clinical severity in sickle cell anemiaBr J Haematol 2005 129:465-81. [Google Scholar]
[2]. Pagnier J, Mears JG, Dunda-Belkhodja O, Schaefer-Rego KE, Beldjord C, Nagel RL, Evidence for the multicentric origin of the sickle cell haemoglobin gene in AfricaProc Natl Acad Sci USA 1984 81:1771-73. [Google Scholar]
[3]. Antonarakis SE, Boehm CD, Serjeant GR, Theisen CE, Dover GJ, Kazazian HH, Origin of the βS-globin gene in Blacks: The contribution of recurrent mutation or gene conversion or bothProc Natl Acad Sci USA 1984 81:853-56. [Google Scholar]
[4]. Serjeant GR, The geography of sickle cell disease: opportunities for understanding its diversityAnn Saudi Med 1994 14:237-46. [Google Scholar]
[5]. Labie D, Pagnier J, Lapoumeroulie C, Rouabhi F, Dunda-Belkhodja O, Chardin P, Common haplotype dependency of high Gγ-globin gene expression and high Hb F levels in β-thalassemia and sickle cell anemia patientsProc Natl Acad Sci USA 1985 82:2111-14. [Google Scholar]
[6]. Month SR, Wood RW, Trifillis PT, Orchowski PJ, Sharon B, Ballas SK, Analysis of the 5’ flanking regions of the gamma globin genes from major African haplotype backgrounds associated with sickle cell diseaseJ Clin Invest 1990 85:364-70. [Google Scholar]
[7]. Kulozik AE, Kar BC, Satapathy RK, Serjeant BE, Serjeant GR, Weatherall DJ, Fetalhemoglobin levels and βS globin haplotype in an Indian population with sickle cell diseaseBlood 1987 69:1742-46. [Google Scholar]
[8]. Sebastiani P, Wang L, Nolan VG, Melista E, Ma Q, Baldwin CT, Fetal hemoglobin in sickle cell anemia: Bayesian modeling of genetic associationsAm J Hematol 2008 83:189-95. [Google Scholar]
[9]. Labie D, Dunda-Belkhodja O, Rouabhi F, Pagnier J, Ragusa A, Nagel RL, The -158 site 5’ to the G gamma gene and G gamma expressionBlood 1985 66:1463-65. [Google Scholar]
[10]. Miller BA, Salameh M, Ahmed M, Wainscoat J, Antognetti G, Orkin S, High fetal hemoglobin production in sickle cell anemia in the eastern province of Saudi Arabia is genetically determinedBlood 1986 67:1404-10. [Google Scholar]
[11]. Nagel RL, Fabry ME, Pagnier J, Zohoun I, Wajcman H, Baudin V, Hematologically and genetically distinct forms of sickle cell anemia in Africa: the Senegal type and the Benin typeN Engl J Med 1985 312:880-84. [Google Scholar]
[12]. Powars D, Hiti A, Sickle cell anemia. Beta S gene cluster haplotypes as genetic markers for severe disease expressionAm J Dis Child 1993 147:1197-1202. [Google Scholar]
[13]. Das SK, Talukder G, Beta globin gene and related: A reviewInt J Hum Genet 2002 3:139-52. [Google Scholar]
[14]. Lehman H, Cutbush M, Sickle cell trait in Southern IndiaBr Med J 1952 1:289-90. [Google Scholar]
[15]. Shukla RN, Solanki BR, Sickle cell trait in Central IndiaThe Lancet 1958 1:297-98. [Google Scholar]
[16]. Kar BC, Devi S, Dash KC, Das M, The sickle cell gene is widespread in IndiaTrans R Soc Trop Med Hyg 1987 81:273-75. [Google Scholar]
[17]. Sutton M, Bouhassira EE, Nagel RL, Polymerase chain reaction amplification applied to the determination of beta-like globin gene cluster haplotypesAm J Hematol 1989 32:66-69. [Google Scholar]
[18]. Varawalla NY, Fitches AC, Old JM, Analysis of beta-globin gene haplotypes in Asian Indians: origin and spread of beta-thalassaemia on the Indian subcontinentHum Genet 1992 90:443-49. [Google Scholar]
[19]. Semenza GL, Dowling CE, Kazazian HH Jr, Hinf I polymorphisms 3’ to the human β globin gene detected by the polymerase chain reaction (PCR)Nucl Acids Res 1989 17:2376 [Google Scholar]
[20]. Niranjan Y, Chandak GR, Veerraju P, Singh L, Some atypical and rare sickle cell gene haplotypes in populations of Andhra Pradesh, IndiaHum Biol 1999 71:333-40. [Google Scholar]
[21]. Mahesh KSSU, Aggarwal A, Bhasker MV, Mukhopadhyay R, Saraswathy KN, Distribution pattern of HbS and β-globin gene haplotypes among Koya Dora tribe of Andhra PradeshInt J Hum Gene 2011 2:123-26. [Google Scholar]
[22]. Mukherjee MB, Surve RR, Gangakhedkar RR, Ghosh K, Colah RB, Mohanty D, Beta-globin gene cluster haplotypes linked to the βS gene in western IndiaHemoglobin 2004 2:157-61. [Google Scholar]
[23]. Labie D, Srinivas R, Dunda O, Dode C, Lapoumeroulie C, Devi V, Haplotypes in tribal Indians bearing the sickle gene: evidence for the unicentric origin of the beta S mutation and the unicentric origin of the tribal populations of IndiaHum Biol 1989 4:479-91. [Google Scholar]
[24]. Bhagat S, Patra PK, Thakur AS, Association between XmnI polymorphism and HbF level in sickle cell disease patients from ChhattisgarhInt J Biomed Sci 2012 1:36-39. [Google Scholar]
[25]. Thein SL, Menzel S, Lathrop M, Garner C, Control of fetal hemoglobin: new insight emerging from genomics and clinical implicationsHum Mol Genet 2009 18:R216-R23. [Google Scholar]
[26]. Lanclos KD, Oner C, Dimovski AJ, Gu YC, Huisman TH, Sequence variations in the 5’ flanking and IVS-II regions of the G gamma-and A gamma-globin genes of beta S chromosomes with five different haplotypesBlood 1991 77:2488-96. [Google Scholar]