Calcineurin inhibitor induced thrombotic microangiopathy is a rare but well recognized complication of a renal transplantation that occurs in 1% of the patients who are on tacrolimus immunosuppression. Among the other aetiological factors of the “de-novo” Thrombotic Microangiopathy (TMA), the condition especially has to be differentiated from an antibody mediated rejection, as both have different pathogenesis, therapeutic connotations and outcomes.
We report a case of a middle aged female renal transplant recipient treated with tacrolimus, who developed localised thrombotic microangiopathy in the early post transplantation period. Despite the normal trough levels of tacrolimus, a diagnosis of “Tacrolimus induced TMA” was rendered after excluding other causes of the “de-novo” TMA, which included an antibody mediated rejection, a meticulous clinico-pathological correlation and serological studies. The treatment included the substitution of tacrolimus by rapamycin, with the subsequent normalization of the renal function.
Calcineurin inhibitors, Renal transplantation, Tacrolimus, Thrombotic microangiopathy
INTRODUCTION
Post renal transplant thrombotic microangiopathy (PT-TMA) is a “recurrent” or “denovo” ,“localized” or a “systemic” devastating disorder that, as per the USRDS data analysis, occurs with an incidence of 5-6 episodes per 1000 people years [1, 2].
Among the proposed aetiological factors, Calcineurin inhibitor induced TMA (CNI-TMA) is a rare but well documented cause of acute renal failure [3]. An early allograft biopsy with a prompt diagnosis of the condition, its distinction from an antibody mediated rejection and its management by drug substitution, plays a vital role in the allograft outcome.
We report a case of localized tacrolimus associated TMA occurring in the early stage after renal transplantation.
CASE REPORT
A 52 year-old Caucasian female underwent a living, non-related, donor transplantation for ESRD. After an initial induction with basiliximab, the post transplant immunosuppression consisted of methylprednisolone (10 mg/day), mycophenolate mofetil (1g/ day) and tacrolimus (5 mg/day). A clinical improvement with good diuresis was achieved for 48 hours, following which the renal functions deteriorated, with a reduction in the urine output and peak serum creatinine levels of 3.8 mg/dl. The laboratory investigations (before dialysis) showed the following: haemoglobin -11.1 g/dl, platelets -1.64 lakhs/mm3, blood urea nitrogen -62 mg/dl, sodium -136 mEq/l, potassium -5.9 mEq/l, chloride -117 mEq/l, phosphate -4.9 mEq/l, uric acid -8.9 mg/dl, complement C3c- 95mgm/dl, C4- 25mgm/dl and lactate dehydrogenase -210 u/l. With a clinical suspicion of acute tubular necrosis/acute rejection, a renal biopsy was done, which revealed fibrin thrombi in the glomerular capillaries [Table/Fig-1 A and B] and arterioles at the glomerular vascular pole with associated fibrinoid necrosis, in a focal and segmental fashion [Table/Fig-2 A and B]. Some of the glomeruli exhibited tuft collapse and widening of the Bowman space. The proximal tubular epithelial cells showed isometric cytoplasmic vacuolization which was suggestive of tacrolimus induced toxic tubulopathy [Table/Fig-3]. A provisional diagnosis of “Thrombotic microangiopathy – Antibody mediated rejection? Tacrolimus induced?” was rendered. Further investigations were done. IHC showed no deposits of C4d in the peritubular capillaries (C4d0 Negative) [Table/Fig-4]. The donor specific antibody levels were < 1000 MFI, the Tacrolimus trough level was 10.1 ng/ml and the tests for double stranded DNA, anticardiolipin antibodies and lupus anticoagulant were negative. The pretransplant work up had revealed a negative viral serology for the HBsAg, antiHCV and the antiHIV antibodies, and other bacterial and viral causes of Thrombotic Microangiopathy (TMA) were ruled out. Repeated peripheral smears showed no evidence of hemolysis, suggesting “localised” TMA. A final diagnosis of “Tacrolimus associated Localised Thrombotic Microangiopathy” was made. Tacrolimus was substituted by mTOR inhibitor rapamycin (a loading dose of 6 mg orally and a subsequent daily dose of 2 mg orally), with subsequent normalization of the renal function.
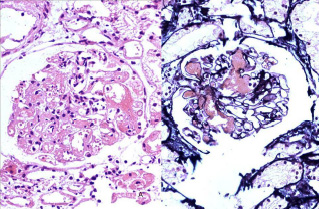
(A) Fibrin thrombi in glomerular capillaries, widening of Bowmans space. H&E X 400
(B) Fibrin thrombi in glomerular capillaries, widening of Bowmans space. Silver methenamine X 400
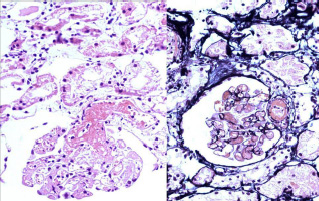
(A) Fibrin thrombi in glomerular capillaries and arterioles at the glomerular vascular pole. H&E X 400
(B) Fibrinoid necrosis and platelet fibrin thrombus affecting arterioles at glomerular vascular pole. Silver methenamine X 400
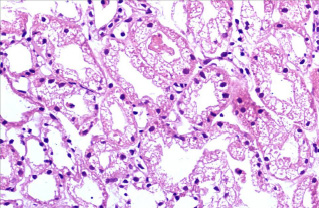
Isometric vacuolization of proximal tubular epithelial cell cytoplasm. H&E X 400
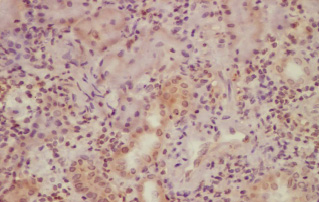
Immunohistochemistry : C4d not detected along peritubular capillaries. Anti-C4d-horseradish peroxidase X 400
DISCUSSION
Thrombotic Microangiopathy (TMA) denotes non-inflammatory small vessel vasculopathies which result from severe endothelial cell injury/ necrosis that culminates in variable degree of organ ischaemia [4,5]. The pathological diagnosis of TMA requires the presence of one or more of the following i) microvascular thrombi ii) a glomerular capil lary occlusion which results from subendothelial deposits of the electronlucent material and iii) a severe subendothelial widening [4,6].
TMA is relatively more common in transplant patients than in the general population, presumably due to the clustering of the risk factors [1]. Post-Transplant TMA (PT-TMA) can be “recurrent”, occurring in patients with a previous history of HUS/TTP or it may be “de-novo”, occurring for the first time post-transplantation [7,8]. The USRDS data analysis quoted the incidence of “de-novo” TMA as 0.8%; however, various studies have reported its incidence to range from 4-14% [8]. This This condition can be triggered by renal ischaemia, antibody mediated rejection (AMR), malignancy, viral infections (CMV, Influenza A, parvovirus B19, BK polyoma virus, HIV and HHV-6), anti-phospholipid antibodies, anti-cardiolipin antibodies in HCV-positive patients and drugs (mTOR inhibitors, antiviral agents) and complicates patients who are on Calcineurin Inhibitors (CNI), affecting 3 to 14% of the patients who are on cyclosporine and approximately 1% of the patients who are on tacrolimus (FK560) [3–5,8,9]. The precise mechanism of CNI induced TMA is not clear; however, it is attributed to the vasoconstrictor , endothelial toxicity, prothrombotic and antifibrinolytic effects of the drug [5,7,8,10]. The trough levels of FK560 are not predictive of developing TMA, as in the present case, and the rising serum creatinine levels may be the only alteration reflecting renal dysfunction.
The clinical presentation may be variable. Some patients present with”localised” TMA manifested by only worsening of renal function, as in the present case , and others present with features of “systemic” TMA which is characterized by microangiopathic haemolysis and thrombocytopenia.
In the present case, after the diagnosis of TMA, the priority was to distinguish the FK560 induced toxicity and AMR as the viral and bacterial causes of TMA, SLE, anti-phospholipid antibody syndrome, scleroderma and recurrent HUS which was usually systemic, were excluded.
The patients with the localized CNI induced TMA often respond to the reduction, temporary discontinuation or the conversion of CNI, usually without a plasma exchange, unlike AMR induced TMA which requires one or more alternatives like a plasma exchange, an intravenous immunoglobulin and the anti-CD20 antibody. Thus, differentiating these two entities which have entirely different pathogeneses and outcomes, is of utmost importance. A distinction is not possible on the basis of the morphology alone. The peritubular capillary C4d positivity is present in AMR and it is a useful discriminator; however, the absence of a C4d staining does not exclude AMR, as 10% of the cases are C4d negative [5,11]. These cases require additional serological studies (anti HLA-donor antibodies) and a meticulous clinicopathological correlation. An irregular intimal proliferation with a subendothelial neutrophilic and a mononuclear infiltration, peritubular capillaritis and an arterial fibrinoid necrosis, with a general involvement of the entire vascular tree, are the features of AMR, whereas the fibrin thrombi which focally affect the arterioles at the glomerular vascular poles and distally extend into the glomerular capillaries in a segmental fashion, suggest CNI induced TMA [7,8,11]. In the current case, the presence of the latter feature along with proximal tubular isometric vacuolization suggested CIN induced TMA, which was further confirmed by a negative C4d staining, low DSA (donor specific antibodies) levels and an improved renal function following the withdrawal of Tacrolimus. A proximal tubular isometric vacuolization in the post-transplant setting should be interpreted with caution, as it may be found in osmotic nephrosis [10].
In conclusion, an early and a prompt diagnosis of CNI induced TMA, it’s further categorization as “localized” and “systemic” and a diligent exclusion of other cases of de-novo TMA, especially AMR, plays a critical role in the allograft outcome.
[1]. Qutube S, Arun KG, Jayaram N, Ramakrishna S, Dilip R, Post transplant thrombotic microangiopathy causing acute renal failureIndian J Nephrol 2009 19(2):74-76. [Google Scholar]
[2]. Reynolds JC, Agodoa LY, Yuan CM, Abbott KC, Thrombotic microangiopathy after renal transplantation in the United StatesAm J Kidney Dis. 2003 42:1058 [Google Scholar]
[3]. Herman MT, Lugan DT, Stephen B, Juan MG, Wadi NS, FK506- Associated Thrombotic microangiopathyTransplantation 1999 67(4):539-44. [Google Scholar]
[4]. Ardalan MR, Review of Thrombotic Microangiopathy (TMA), and Post- Renal Transplant TMASaudi J Kidney Dis Transpl. 2006 17:235-44. [Google Scholar]
[5]. Nickeleit V, Thrombotic Microangiopathy in Renal Allografts. [monograph on the Internet]. Chapel Hill. Available from:http://www.nephropathology-esp.org/uploads/user-3/lectures/ecp-istanbul-2007/tma_tx_istanbul07.pdf [Google Scholar]
[6]. Schwimmer J, Nadasdy TA, Spitalnik PF, Kaplan KL, Zand MS, De novo thrombotic microangiopathy in renal transplant recipients: A comparison of haemolytic uremic syndrome with localized renal thrombotic microangiopathyAm J Kidney Dis. 2003 41:471-79. [Google Scholar]
[7]. Norisa M, Remuzzia G, Thrombotic Microangiopathy After Kidney TransplantationAmerican Journal of Transplantation 2010 10:1517-23. [Google Scholar]
[8]. Ponticelli C, Banfi G, Thrombotic microangiopathy after kidney transplantationTransplant Int. 2006 19(10):789-94. [Google Scholar]
[9]. Kais H, Nourredine C, Raoudha B, Emira C, Tarek S, Fehmi S, Treatment of Tacrolimus-associated thrombotic microangiopathy in renal transplant recipient with fresh frozen plasma: A case reportSaudi J Kidney Dis Transpl 2006 17:58-61. [Google Scholar]
[10]. Maarten N, Dirk RJK, Minnie S, Calcineurin Inhibitor NephrotoxicityClin J Am Soc Nephrol 2009 4:481-509. [Google Scholar]
[11]. Colvin BR, Nickeleit V, Renal Transplant Pathology. In: Jennette CJ, Olson LJ, Schwartz MM, Silva GF, editorsHeptinstall’s Pathology of the Kidney 2007 6thPhiladelphiaLippincott Willams and Wilkins:1347-490. [Google Scholar]