Lipoid Proteinosis (LP) is a genetically linked, autosomally transferred, rare, chronic multisystem disease which is characterized by a normal lipid profile, but with abnormal deposits of lipids and proteins in the body, which slowly but steadily leads to systemic manifestations. Although it affects almost all the systems of the body, it predominantly manifests as lesions on the skin and it has characteristic intracranial calcifications. Although, the intracranial calcifications can be classified, based on their aetiopathogenesis, as agerelated and physiologic, congenital, infectious, endocrine and metabolic, vascular, and neoplastic; the symmetric calcifications in LP are a distinct entity. To one who is aware of this entity, LP is usually an incidental diagnosis. No permanent cure is available for LP till date. Only symptomatic medical treatment is being given. With the increasing awareness on this entity, LP can now be detected in its early phase and it can be better managed.
As this condition is rare, it is necessary to spread awareness on this entity in the scientific community and hence this case is being reported. This case report is the first to demonstrate a novel association of an additional intracranial calcification in Lipoid Proteinosis.
Introduction
Lipoid Proteinosis (LP) is a radiologically rare genodermatosis which is caused by the abnormal deposition of lipids and proteins in various systems of the body, even though the lipid profile normal [1,2].
Although multiple systems of the human body are affected by it, the most predominant manifestations which are seen in the dermatology practice are lipid laden skin lesions and eye lesions and in the radiology practice, symmetric, intracranial, medial, temporal calcifications are seen. To one who is aware of this entity, LP is usually an incidental diagnosis. As this condition is rare, it is necessary to spread awareness on this entity in the scientific community. Hence, a case of LP which was diagnosed incidentally in a 52 years male who was referred for CT scan of the head with complaints of occasional headache has been reported here.
Case Report
A 52 years male was referred for CT scan examination of the head for headache that was troubling him for the past 6 months.
Clinically, he was conscious, co-operative and well oriented to the time, place and persons. He had no prior history of any systemic diseases. Yellowish patches [Table/Fig-1] were seen on his skin, over the forearm. There was absence of any hyperkeratosis of the skin and depigmentation of the lips. The tongue was freely mobile. There was no hoarseness of the voice.
Yellowish patches are seen on the skin over the left forearm
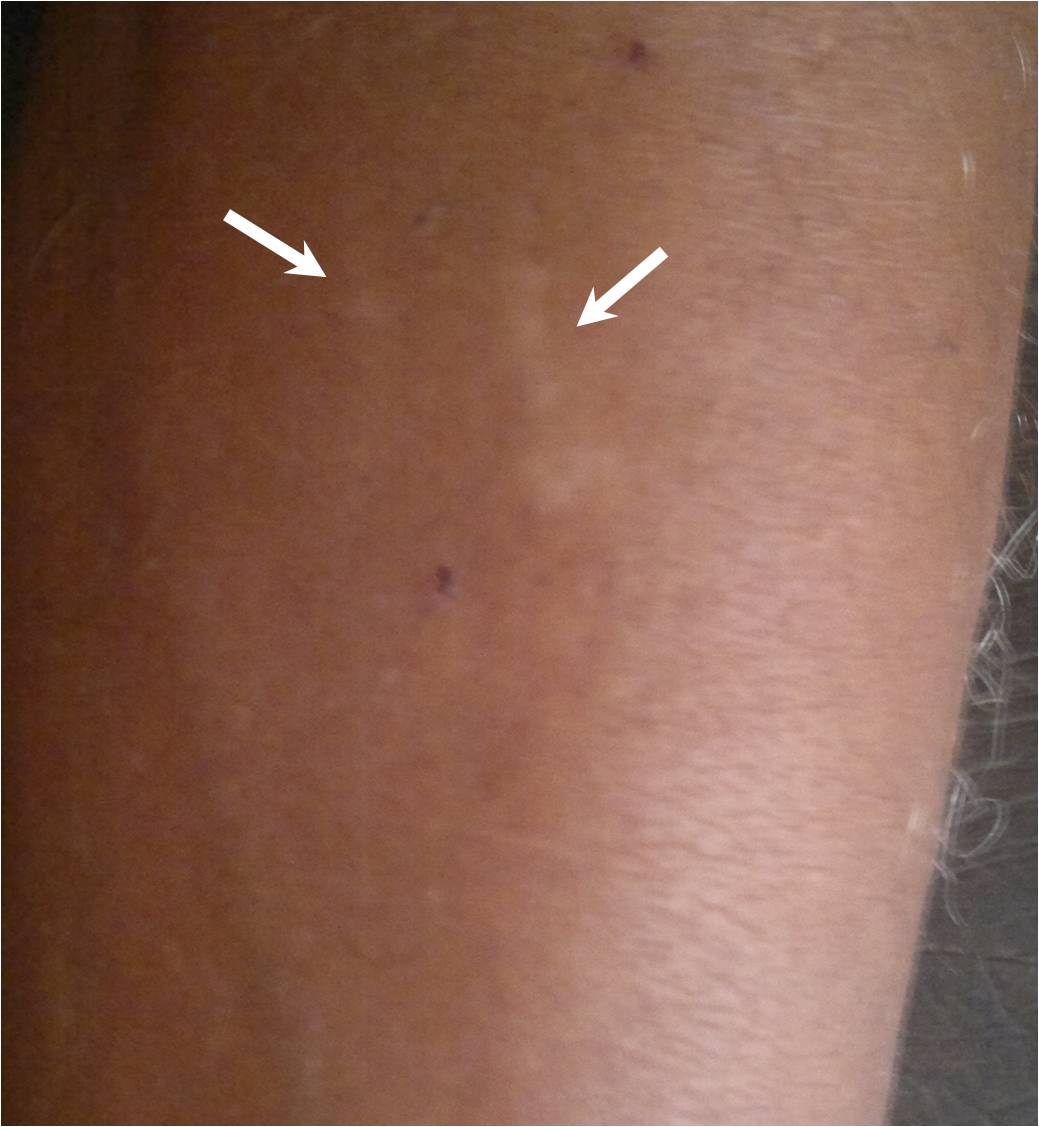
There was neither any significant family history, nor was he a product of a consanguineous marriage.
His blood counts and lipid profile were within normal limits. His serum cholesterol was 144 mg/dl (normal < 200 mg/dl), his serum triglycerides were 96 mg/dl (normal < 170 mg/dl) and his serum HDL cholesterol was 53 mg/dl (normal < 70 mg/dl).
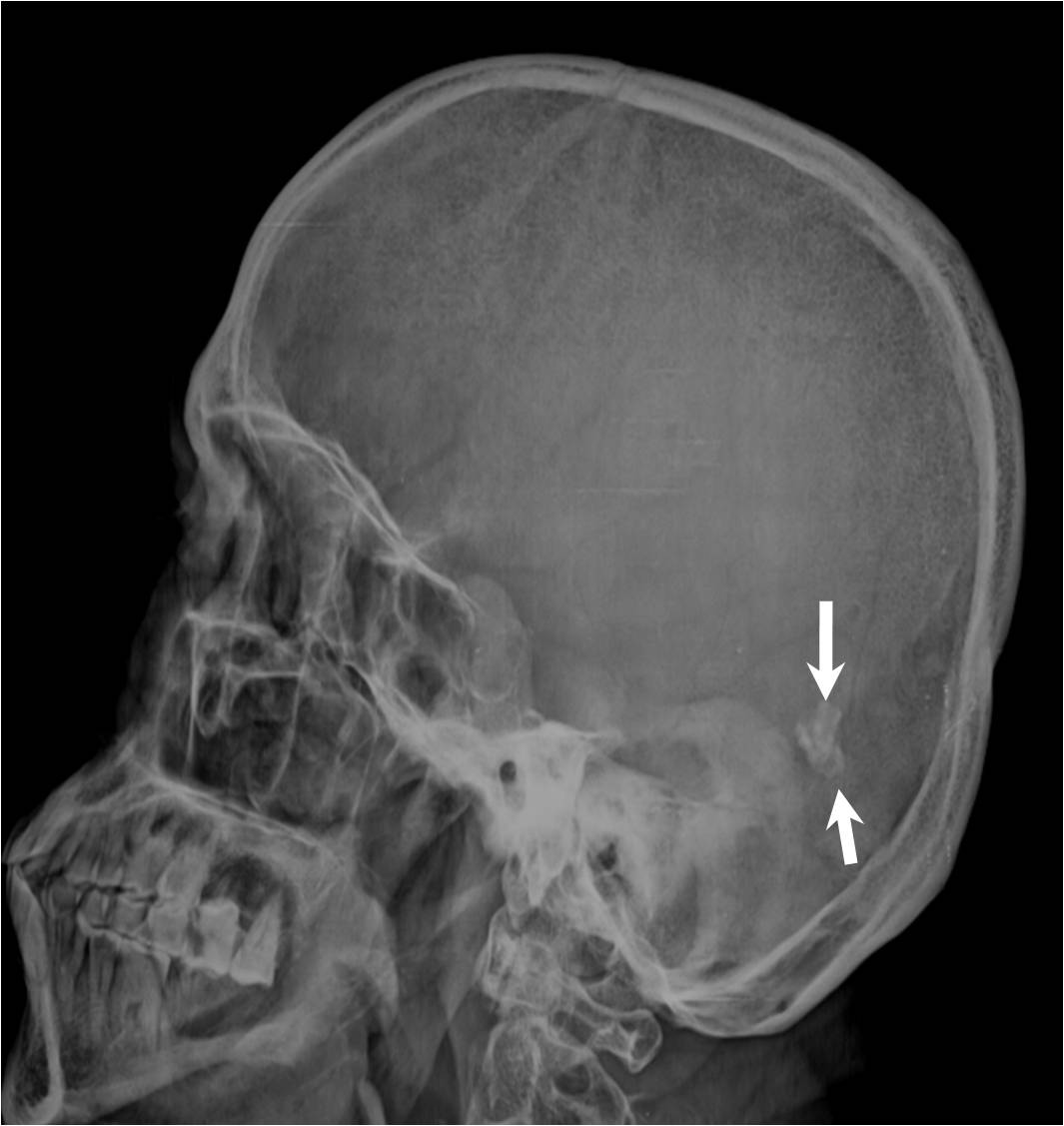
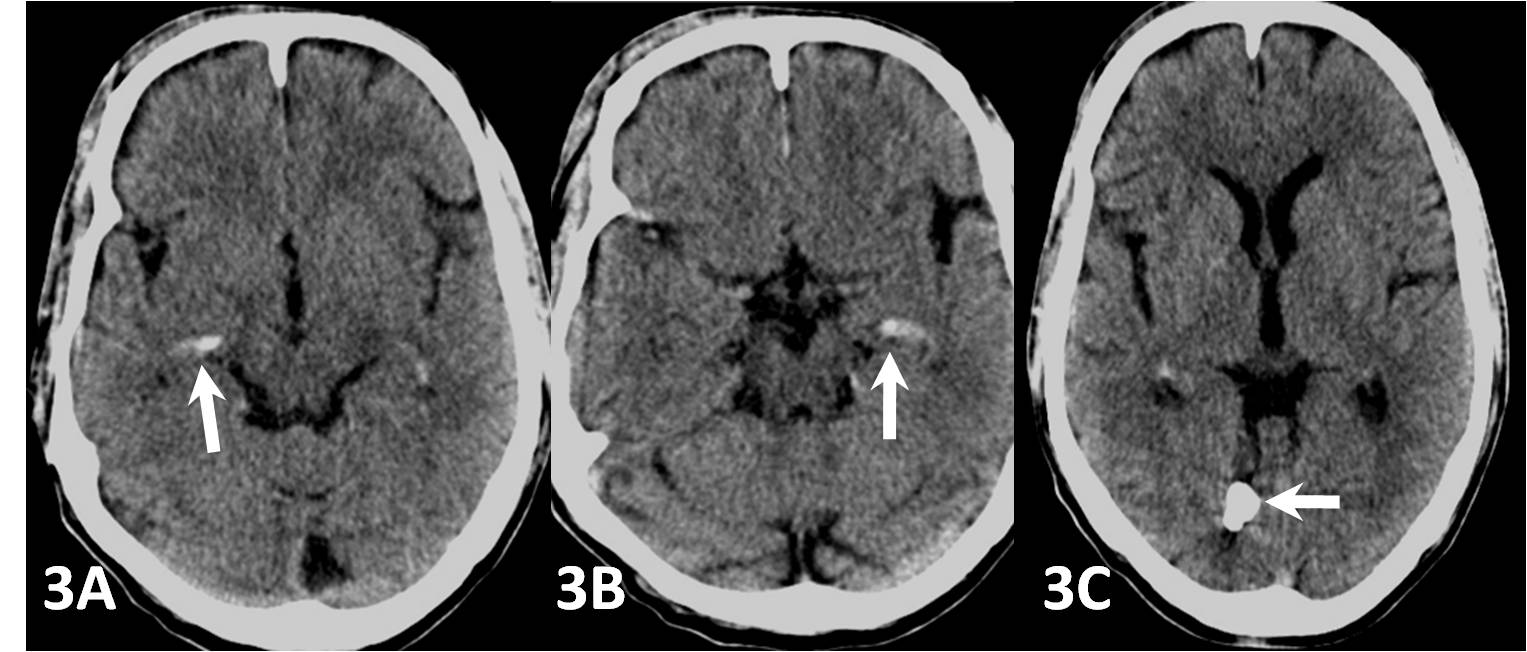
The plain radiograph of the skull in the lateral view [Table/Fig-2] showed an irregular intracranial calcification in the posterior cranial fossa region. The plain CT scan brain window images showed subtle, symmetric, comma shaped hyperdensities with a CT attenuation value of 84 – 92 H.U. which were suggestive of calcifications in the medial aspect of the temporal lobes [Table/Fig-3(a), 3(b)], in addition to the irregularly shaped, posterior fossa intracranial calcification [Table/Fig-3(c)]. Hence, a radiological diagnosis of LP was put forth.
Lateral skull radiograph shows intracranial calcification in posterior fossa
Plain CT scan (brain window images) shows subtle symmetric comma shaped calcifications in medial aspect of the temporal lobes (3A and 3B) classically seen in LP and additional calcifications in posterior fossa (3C) which is the novelty in this case.
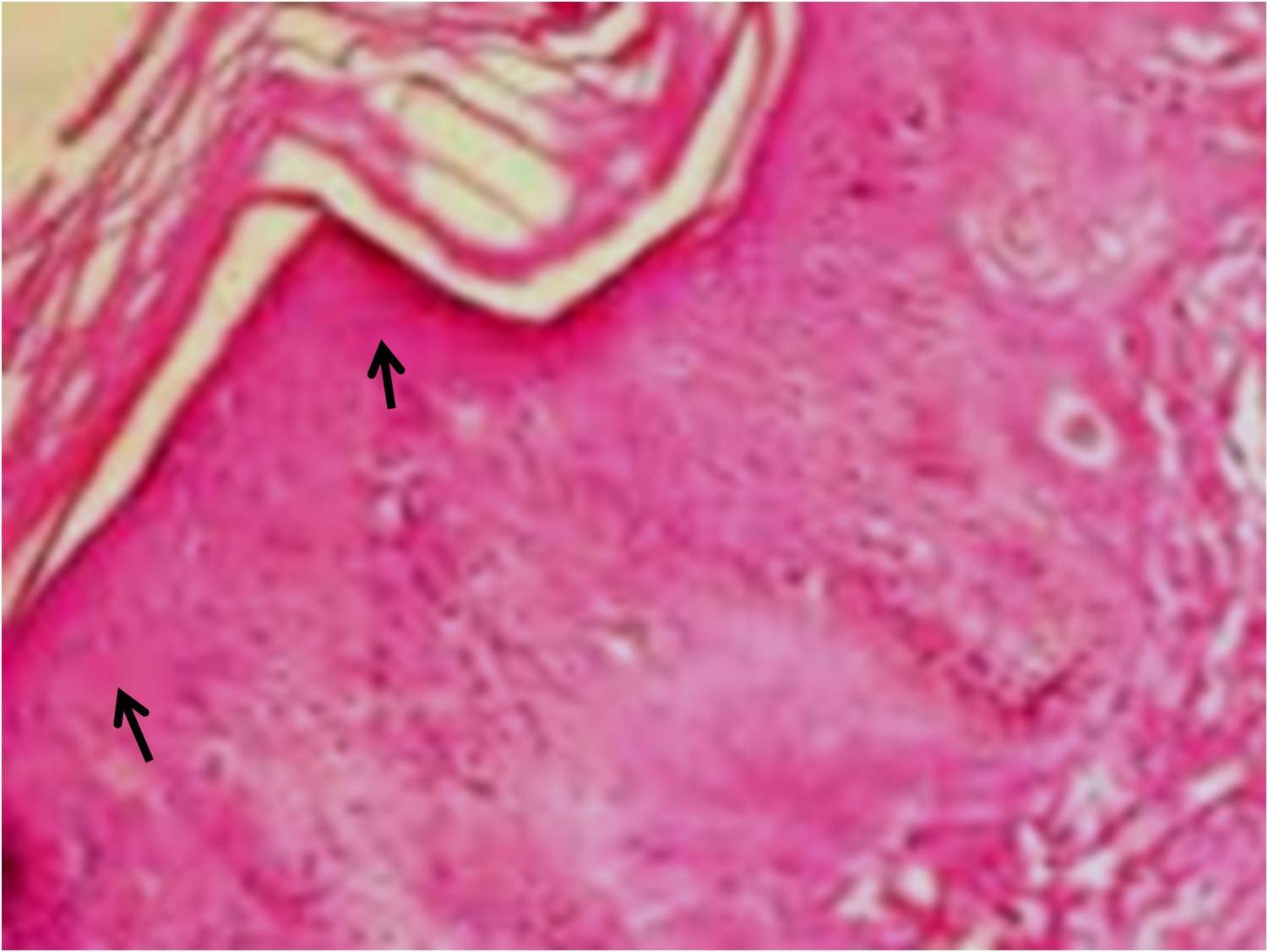
The histopathology of the punch biopsy from the yellowish plaques on the skin which was done outside by the referring physician, reported them as ‘an abundant deposition of amorphous eosinophilic material in the thickened papillary dermis.’ The hyaline like material was diastase resistant and PAS positive [Table/Fig-4]. These findings were consistent with the radiological findings of LP.
Histopathology of punch biopsy from yellowish plaques on skin showed abundant deposition of amorphous PAS positive eosinophilic material surrounding the sweat glands
Discussion
Although Seibman first reported Lipoid Proteinosis in 1908 [3], Urbach and Weithe scientifically studied it and named it as “Lipoidosis cutis et mucosae” in 1929 [3,4].
Hamada et al. [5] mapped LP to chromosome 1q21 at D1S498 [6] and concluded that the mutations outside the exon 7 exhibited the more severe mucocutaneous LP phenotype [7].
LP has a slow but steady and a progressive course. Histologically, the material which is deposited in the tissues resembles lipids and proteins. Hence, this entity was erroneously labeled as LP.
Very few cases have been reported from Asia and most of the cases have been seen in South Africa and Central Europe [3,8]. Such is the rarity of the disease that till date, not more than 500 cases have been reported worldwide. Therefore, the actual picture on its incidence and prevalence is not completely known.
No definitive age, sex or race predilections exist. There is a documented autosomal recessive inheritance as there is usually a history of consanguinity among the unaffected parents [9]. A mental sub normality is seen only when it is transmitted by autosomal dominant inheritance due to the mutant genes [3].
In the gene which encodes the extracellular matrix protein 1 (ECM1) on the chromosome band 1q21, loss of function mutations occur. The patients with the exon 7 mutations display slightly milder clinical features, while the mutations in exon 6 result in a more severe phenotype [5,6]. This data continues to get modified and recently, a novel nonsense mutation in the ECM1 gene was identified in a Pakistani family [7].
A glycoprotein which is produced by the normal ECM1 gene is expressed in the skin, mucosa and in multiple internal organs. The mutations in this gene lead to the deposition of a hyaline like material in the skin and the viscera in abnormal amounts, which is the cause of the clinical manifestations. These deposits stain positive with the Periodic Acid-Schiff stain and they are Diastase resistant and negative for Congo red [10].
Due to the deposition of this hyaline like material in the oesophagus, a barium swallow examination shows filling defects in the upper aero-digestive tract. The most common radiological hallmark is the presence of bean or comma shaped intracranial calcifications in the temporal lobes in the amygdala, which is more evident in the patients who have LP for a long duration. Epilepsy, when it is present, may be related to these calcifications. Hence, a complete evaluation which is done by performing an MRI/CT scan in the patients with LP is recommended, in order to identify these abnormalities [11]. The radiographs and CT are adequate for a definitive diagnosis, while MRI is useful for assessing the brain in totality. The case which has been reported here showed medial temporal calcifications which were typical of LP and an additional bizarre calcification in the right posterior parietal region, which has never been reported earlier.
Radiologically, such typical intracranial calcifications are rare in other disorders in the presence of a normal biochemical profile. Although the skin lesions in Erythropoietic Protoporphyria (EPP) have a similar appearance, the deposits are not seen around the sweat glands. Increased protoporphyrin levels in erythrocytes are also seen in EPP [11] as in LP. The deposits in amyloidosis and xanthomas have different chemical compositions; although externally, the skin might appear similar as in LP.
LP is not always asymptomatic. With a protracted course; the complications of LP are: airway obstruction due to the laryngeal involvement, hoarseness of the voice and impaired speech due to the vocal cord involvement, gastrointestinal bleeding if the small bowel is involved and corneal opacities and secondary glaucoma which are due to the ocular involvement [3,5].
There is no permanent cure for LP. The symptomatic medical treatment for the skin lesions includes dimethyl sulphoxide, etretinate, intralesional heparin and D-penicillamine. However, none of these medicines have shown consistently good results. Dermabrasion for the skin lesions is done in the cases which show no improvement by the oral medications. Carbon dioxide laser for the lesions of the eyelids and the aero digestive tract is also under consideration. Anticonvulsants are used when the patient has seizures [1,2].
To conclude, although LP is not always a life threatening entity, it can at times be quite distressing. An appropriate radio-pathological workup can lead to an early detection so that a team effort of multiple specialties can render a proper management.
The novelty in the case which has been reported here, is the association of the posterior fossa calcification in addition to the routinely seen medial temporal calcification of LP.
[1]. Kachewar S, Singh H, Sasane AG, Bhadane S, A full blown case of lipoid proteinosis. A case reportMJAFI 2011 67:90-91. [Google Scholar]
[2]. Kachewar S, Singh H, Bhadane S, Khandelwal A, Pawar A, Lipoid Proteinosis. A review articleNepal Journal of Neuroscience 2009 6:5-8. [Google Scholar]
[3]. Sainani MP, Muralidhar R, Parthiban K, Vijayalakshmi P, Lipoid proteinosis of Urbach and Weithe: a case report and a brief review of the literatureInt Ophthalmol. 2011 Apr 31(2):141-43. [Google Scholar]
[4]. Urbach E, Wiethe C, Lipoidosis cutis mucosaeVirchows Arch Patholog Anat 1929 273:285-319. [Google Scholar]
[5]. Hamada T, McLean WHI, Ramsay M, Ashton GHS, Nanda A, Jenkins T, Lipoid proteinosis maps to chromosome 1q21 and it is caused by mutations in the extracellular matrix protein 1 gene (ECM1)Hum Molec Genet 2002 11:833-40. [Google Scholar]
[6]. Hamada T, Wessagowit V, South AP, Ashton GHS, Chan I, Oyama N, Siriwattana A, Extracellular matrix protein 1 gene (ECM1) mutations in lipoid proteinosis and the genotype phenotype correlationJ Invest Derm 2003 120:345-50. [Google Scholar]
[7]. Nasir M, Latif A, Ajmal M, Qamar R, Naeem M, Hameed A, The molecular analysis of lipoid proteinosis: identification of a novel nonsense mutation in the ECM1 gene in a Pakistani familyDiagn Pathol. 2011 6:69PMID: 21791056 [Google Scholar]
[8]. Callizo M, Ibáñez-Flores N, Laue J, Cuadrado V, Graell X, Sancho JM, Eyelid lesions in lipoid proteinosis or Urbach-Wiethe disease: a case report and review of the literatureOrbit. 2011 Oct 30(5):242-4.[PMID: 21957955] [Google Scholar]
[9]. Samdani AJ, Azhar A, Shahid SM, Nawab SN, Shaikh R, Qader SA, A homozygous frame shift mutation in the ECM1 gene in two siblings with lipoid proteinosisJ Dermatol Case Rep. 2010 Dec 31 4(4):66-70.[PMID:21886756] [Google Scholar]
[10]. Salih MA, Abu-Amero KK, Alrasheed S, Alorainy IA, Liu L, McGrath JA, Molecular and neurological characterizations of three Saudi families with lipoid proteinosisBMC Med Genet. 2011 Feb 24 12:31[PMID: 21349189] [Google Scholar]
[11]. Ozbek SS, Akyar S, Turgay M, Computed tomography findings in lipoid proteinosis: a report of two casesBr J Radiol 1994 67:207-09. [Google Scholar]