We are describing two sisters with the rare Senior-Loken syndrome, which is a combination of familial juvenile nephronophthisis and retinal dystrophy. The earliest presenting features include an impaired urinary concentrating ability, leading to polyuria and polydipsia and these are associated with visual impairment. The two patients had blindness shortly after their births. They presented to us with evidence of chronic kidney disease (CKD) in their teens, that required the initiation of the renal replacement therapy. We are reporting these two cases, as this was the first occurrence of this condition in the State of Qatar.
Introduction
The Senior-Loken syndrome is a rare syndrome which consists of nephronophthisis (NPHP) and a retinal lesion. It was first described in 1961 by Senior et al, who described a family with six among 13 children who had nephronophthisis and tapetoretinal degeneration [1]. In the same year, Loken et al described the same condition in two siblings; both the siblings had blindness and severe renal failure; a kidney biopsy showed renal tubular atrophy and dilatation [2].
Nephronophthisis is an autosomal recessive condition which presents with end stage renal disease (ESRD) in childhood. The infantile form has an early onset that progresses to ESRD in early life, whereas the juvenile form usually presents with a decreased urinary concentrating ability which progresses to ESRD during the second decade [3–5].
The retinal lesions in the Senior-Loken syndrome are variable, ranging from severe infantile onset retinal dystrophy to a more typical retinitis pigmentosa [6].
Case Report
We are reporting two female patients, 15- and 17-year olds who were born to first degree consanguineous parents. The two sisters presented in their first year with nystagmus and inability to follow light and objects. There fundus examinations were within normal limits. They had polyuria, polydipsia and nocturia since their young ages. By the age of 12, they were found incidentally to have CKD. They had one elder sister and two younger brothers who were normal. There was no family history of renal diseases, and hearing and visual problems.
On referral to the nephrology clinic, the two patients were found to be pale; they were neither jaundiced nor oedematous. Their blood pressurea were normal. They were blind and had nystagmus. There was no retinal pigmentation or cataract on the fundus examination. They did not have any dysmorphic features. The other systemic examinations were unremarkable. Investigations revealed that CKD was present with elevated blood urea nitrogen and serum creatinine values and mild acidosis. They had normal fasting plasma glucose values which ranged from 4-5 mmol/l and the HBA1c value was less than 5%. Their haemoglobin levels were low (in the range of 8 to 9 g/dl), with normal leukocyte and platelet counts. The peripheral blood smears showed dimorphic (normocytic normochromic and microcytic hypochromic) red blood cells. The sickle cell testing was positive in the younger patients, while haemoglobin electrophoresis showed Hb A- 56% and Hb S- 35%. Urinalysis revealed a low specific gravity (always below 1.005) and a urine osmolality which was below 200 mosm/l in both the patients. There was no hematuria, proteinuria or pyuria.
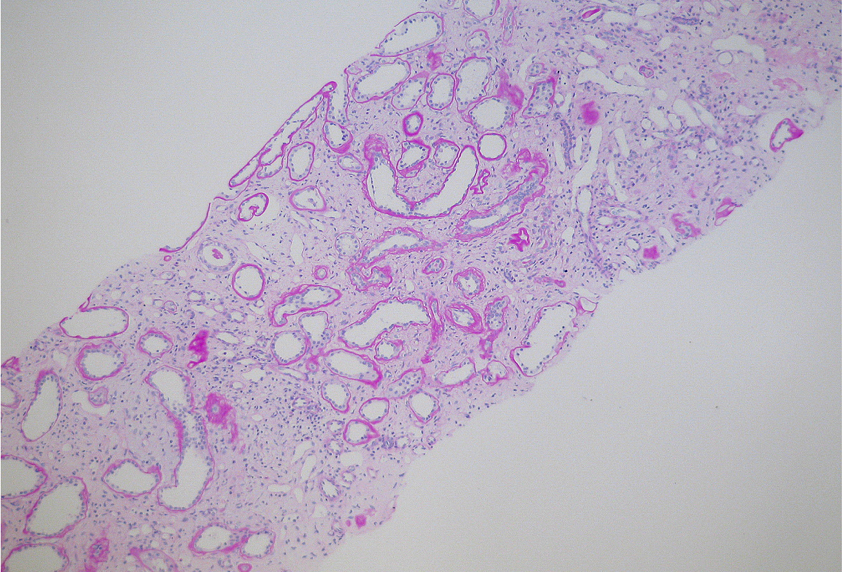
Abdominal ultrasound of the two cases showed undersized hyperechoic kidneys. The liver showed a slightly increased echotexture and the spleen was normal. MRI of the kidneys revealed tiny cysts in the renal parenchyma, which measured 3 mm. The MRI scans of the brain and the eyes of both the patients were normal. The visual evoked response was normal also in both the patients. Kidney biopsies which were performed for both the patients showed moderate chronic nephropathy with some totally sclerosed glomeruli. The remaining showed compensatory hypertrophy. There was moderate tubular atrophy and interstitial fibrosis. Periodic acid-Schiff (PAS) staining showed an irregularly thickened and lamellated renal tubular basement membrane [Table/Fig-1]. The immunofluorescence stain was negative. The histological features were consistent with the clinical impression of nephronophthisis. Unfortunately, a family work-up and genetic testing were not performed.
Kidney biopsy with Periodic acid Schiff (PAS) stain showing interstitial fibrosis with mild focal chronic interstitial inflammation, moderate tubular atrophy and irregular thickening and lamellation of the tubular basement membrane consistent with nephronophthisis
Based on the clinical picture, the early childhood blindness and nephronophthisis, and the pathological findings, the two patients were diagnosed as having the Senior-Loken syndrome. In addition, the younger patient had a sickle cell trait. The two patients were followed up in the Nephrology Clinic at Hamad Medical Corporation, Doha, Qatar. They were started on haemodialysis in our unit once they reached ESRD at the ages of 15 and 17 years respectively. They were dialyzed for 6 months and then they had kidney transplants.
Discussion
The association of nephronophthisis and tapeto-retional degeneration was described by both Senior and Loken in 1961 [1,2]. It occurs in approximately 10-15% of all the cases of nephronophthisis. In addition to the ocular involvement, the clinical and the pathological features of the Senior Loken syndrome are similar to those of isolated nephronophthisis.
The ocular involvement takes several forms: It could be early and severe congenital amaurosis of the Leber type or late-onset pigmentary retinal degeneration which was characterized by night blindness, which was followed later by complete visual loss. Leber’s congenital amaurosis represents a severe form of ocular involvement which leads to blindness in infancy, nystagmus and a diffuse atypical retinal pigmentation with narrowing of the retinal arteries and pallor of the optic disc, with an early and complete extinction of the electroretinogram (ERG) [7]. The tapetoretinal degeneration varies in its nature and severity and it is the commonest form. It is characterized by a progressive degeneration of the choroid and the retina. Retinitis pigmentosa is characterized by bone spicule degeneration that begins from the periphery of the retina and extends to involve the whole of retinochoroid [8]. Retinitis pigmentosa is seen in most of the NPHP genes mutations, but it is more severe in the patients with the NPHP 5 and the NPHP 6 gene mutations [5]. The other ocular findings include cataract, Coat’s disease and keratoconus [9].
The retinal disease may become progressive and so an annual eye examination which commences at the time of the diagnosis is recommended. An electroretinogram (ERG) helps in the diagnosis of these varieties before the retinitis pigmentosa may be observed by a fundoscopic examination.
Nephronophthisis is a disorder which was first described in 1945. It is characterized by chronic tubulointerstitial nephritis that progresses to ESRD during the second decade. To date, the mutations in 9 genes (NPHP1-9) have been identified as the causative agents which are responsible for nephronophthisis [10]. Basically, there are three clinical variants of nephronophthisis according to the age of onset of the ESRD [4].
Firstly, the severe infantile form of nephronophthisis which is associated with blindness in infancy, severe hypertension and death from renal failure before the age of ten. It is caused by the mutations in the NPHP2 gene which is located on chromosome 9q31. It is associated with some extrarenal features which are peculiar to infantile nephronophthisis, which include hypertension, situs inversus, and ventricular septal defects [4,5].
Secondly, the adolescent form is caused by mutations in the NPHP3 gene which is located on chromosome 3q22, in which ESRD occurs in early adulthood and the histological features are similar to those of the juvenile form [4,5].
Thirdly, the juvenile form is the most common type of nephronophthisis and it represents about 5-10% of all the cases of ESRD in children. It affects both the genders equally [4,5]. It is caused by mutations in eight different genes (NPHP 1, 3, 4, 5, 6, 7, 8 and 9) [11]. NPHP1 is the commonest gene (in 25% of the cases) which is responsible for juvenile nephronophthisis. It is characterized by a later onset, a slower progression of the renal disease and milder ocular manifestations. The renal involvement is often asymptomatic and mild. The earliest signs are decreased urinary concentration ability, which leads to polyuria, polydipsia and renal sodium loss by the age of 4-6 years [12]. Proteinuria and haematuria are absent or minimal and the urinary sediment is often normal. The blood pressure remains normal until the development of the chronic renal failure. The disease progresses invariably to CKD before the age of 20 years [3]. However, a late onset of the renal disease in the third and fourth decades was described in two siblings from Greece [13].
The ultrasonographic findings of juvenile nephnonophthisis may be normal or they may show an increased echogenicity of the renal parenchyma, a poor corticomedullary differentiation, a small kidney size, and medullary cysts. However, the lack of medullary cysts at presentation does not rule out the diagnosis of juvenile nephronophthisis [14].
The main histological findings in the kidney biopsies of the juvenile form are moderate to severe tubulointerstitial fibrosis, tubular dilatation and tubular atrophy, with thickening and multilayering of the tubular basement membrane. The glomeruli are basically normal, but a secondary glomerosclerosis can be seen in advanced nephronophthisis. Whereas, the infantile form shows cortical microcysts and a cystic dilatation in the collecting ducts, rather than the thickening and multilayering of the tubular basement membrane that is seen in the juvenile form [5].
Nephronophthisis should not be confused with autosomal dominant polycystic kidney disease (ADPKD) which is characterized by bilateral, multiple renal cysts or medullary cystic kidney disease (MCKD), which shares macroscopic and microscopic features with nephronophthisis. However, unlike nephronophthisis, ADPKD and MCKD are inherited in an autosomal dominant pattern and they progress to ESRD at later ages. The diagnosis of nephronophthisis is based on the genetic testing, the clinical features and the pathological findings [15].
To date, there are no proven treatments for nephronophthisis. The management depends mainly on delaying the progression of the renal failure, which leads to ESRD and the need for dialysis and transplantation. The management in a low clearance renal clinic is appropriate, to allow time for the consideration of renal replacement therapies. Successful renal transplantations have been performed in patients with juvenile nephronophthisis, without the recurrence of the disease in the transplanted organ. Newer agents, vasopressin V2 receptor antagonists, which alter the cystogenesis and the progression of the disease may be available for future use [16].
To the best of our knowledge, this is the first report on the rare Senior Loken syndrome from the state of Qatar. It must be considered in patients who present with visual impairment and renal failure at early ages. There is no specific treatment; however, renal transplantation is the preferred therapy because nephronophthisis will not recur after transplantation.
[1]. Senior B, Friedmann AI, Braudo JL, Juvenile familial nephropathy with tapetoretinal degeneration. A new oculorenal dystrophyAm J Ophthalmol 1961 52:625-33. [Google Scholar]
[2]. Loken AC, Hanssen O, Halvorsen S, Jolster NJ, Hereditary renal dysplasia and blindnessActa Paediatr 1961 50:177-84. [Google Scholar]
[3]. Hildebrandt F, Strahm B, Nothwang HG, Gretz N, Schnieders B, Singh-Sawhney I, Molecular genetic identification of families with juvenile nephronophthisis type 1: the rate of its progression to renal failureKidney Int 1997 51(1):261-9. [Google Scholar]
[4]. Saunier S, Salomon R, Antignac C, NephronophthisisCurr Opin Genet Dev 2005 15(3):324-31. [Google Scholar]
[5]. Salomon R, Saunier S, Niaudet P, NephronophthisisPediatr Nephrol 2009 24:2333-44. [Google Scholar]
[6]. Satran D, Pierpont ME, Dobyns WB, Cerebello-oculo-renal syndromes including Arima, Senior-Loken and the COACH syndromes: More than just variants of Joubert syndromeAm J Med Genet A 1999 86:459-69. [Google Scholar]
[7]. Bell AH, McKiernen PJ, Savage JM, Reid MM, Hereditary renal and retinal dysplasia-the Senior-Loken syndromeUlster Med J 1987 56(2):160-2. [Google Scholar]
[8]. Fillastre JP, Guenel J, Riberi P, Marx P, Whitworth JA, Kunh JM, Senior-Loken syndrome (nephronophthisis and tapeto-retinal degeneration): a study of 8 cases from 5 familiesClin Nephrol 1976 5(1):14-9. [Google Scholar]
[9]. Oyama T, Usui T, Hasebe H, Miki A, Matsumoto S, Suda K, Two cases of Senior-Loken syndromeNippon Ganka Gakkai Zasshi 2004 108(1):29-37. [Google Scholar]
[10]. Otto EA, Tory K, Attanasio M, Zhou W, Chaki M, Paruchuri Y, Hypomorphic mutations in the meckelin (MKS3/TMEM67) cause nephronophthisis with liver fibrosis (NPHP11)Journal of Medical Genetics 2009 46(10):663-70. [Google Scholar]
[11]. Hildebrandt F, Zhou W, Nephronophthisis-associated ciliopathiesJ Am Soc Nephrol 2007 18(6):1855-71. [Google Scholar]
[12]. Gusmano R, Ghiggeri GM, Caridi G, Nephronophthisis-medullary cystic disease: clinical and genetic aspectsJ Nephrol 1998 11(5):224-8. [Google Scholar]
[13]. Bollee G, Fakhouri F, Karras A, Noel LH, Salomon R, Servais A, Nephronophthisis related to homozygous NPHP1 gene deletion as a cause of chronic renal failure in adultsNephrol Dial Transplant 2006 21(9):2660-3. [Google Scholar]
[14]. Blowey DL, Querfeld U, Geary D, Warady BA, Alon U, Ultrasound findings in juvenile nephronophthisisPediatr Nephrol 1996 10(1):22-4. [Google Scholar]
[15]. Simms RJ, Eley L, Sayer JA, NephronophthisisEur J Hum Genet 2009 17(4):406-16. [Google Scholar]
[16]. Gattone VH, 2ndWang X, Harris PC, Torres VE, Inhibition of renal cystic disease development and progression by using a vasopressin V2 receptor antagonistNat Med 2003 9(10):1323-6. [Google Scholar]