Case Report
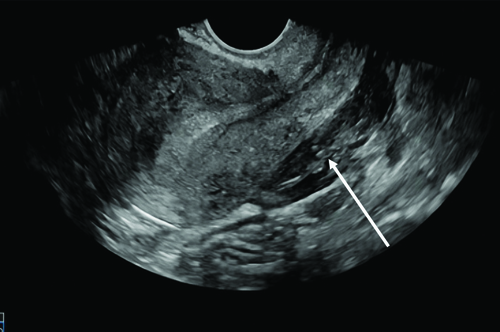
A 29-year-old female homemaker presented to the emergency department with altered mental status for the past three hours. Her vital signs at presentation are listed in [Table/Fig-1], showing cold peripheries, hypotension and absent bowel sounds on auscultation. Extended Focused Assessment Sonography in Trauma (eFAST) suggested a ruptured ovarian cyst with gross haemoperitoneum [Table/Fig-2]. On examination, a malar rash was observed, along with petechiae and purpura all over the body.
Illustrating the vital recordings during presentation.
| Vitals | Findings | Normal |
|---|
| Blood pressure (mmHg) | 80/50 | 120/80 |
| Heart rate (beats per minute) | 120-140 | 60-100 |
| Respiratory rate (beats per minute) | 22-30 | 12-20 |
| Temperature (°C) | 36-37 | 37 |
| SpO2 (%) | 92 | 95-100 |
Ultrasound image depicting gross haemoperitoneum (white arrow).
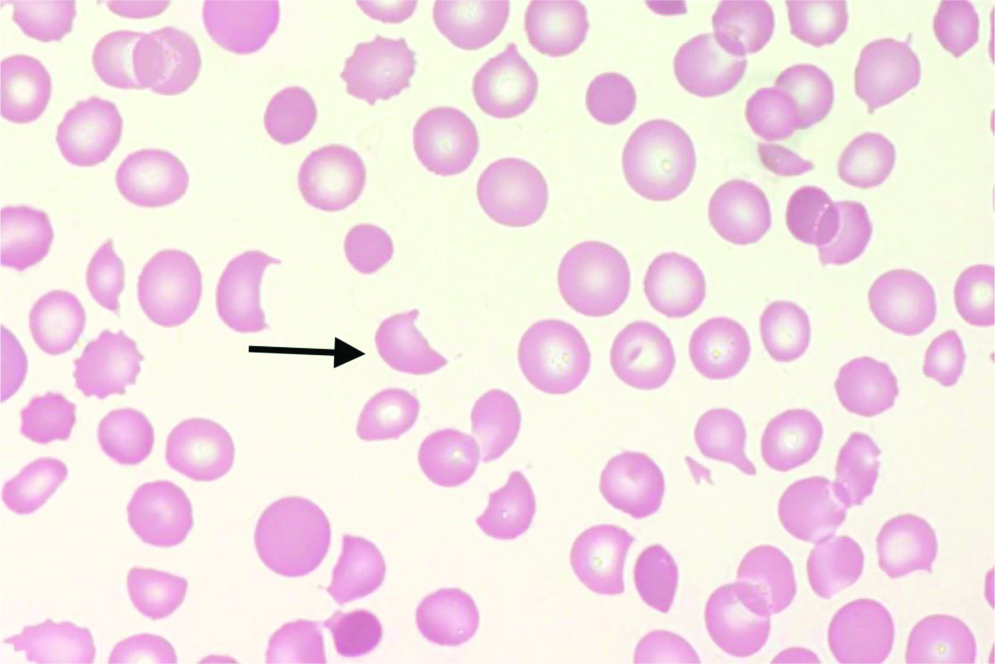
A complete blood count showed severe thrombocytopenia [Table/Fig-3], and the peripheral blood smear demonstrated schistocytes and polychromasia. Further investigation revealed no significant past or family history, drug allergies, or adverse reactions to medications. The patient was immediately resuscitated and taken for emergency laparotomy.
Complete blood picture at the time of presentation.
| Complete blood picture | Findings | Normal range |
|---|
| Haemoglobin (Hb) (g/dL) | 8 | 12.0-15.5 (females) |
| White blood cells (/μL) | 13000 | 4,000-11,000 |
| Platelets (/μL) | 20000 | 150,000-450,000 |
Day by Day Progression and Intervention
Postoperative Day (POD)-0: After an emergency laparotomy, the patient reported nausea and mild localised abdominal tenderness at the surgical site. Despite these complaints, her vitals were stable, suggesting early haemodynamic stability post-surgery. A Complete Blood Count (CBC) after surgery revealed severe thrombocytopenia, with a platelet count of 8,000/μL and Haemoglobin (Hb) of 7.2 mg/dL. A peripheral smear showed anisopoikilocytosis and polychromasia with schistocytes [Table/Fig-4]. The patient received a Single-Donor Platelets (SDP) transfusion, which increased the platelet count to 65,000/μL, providing temporary stabilisation. These haematological findings prompted further investigations to rule out potential causes such as MAHA, TTP, and SLE. Additional investigations were ordered [Table/Fig-5], which showed low Hb and platelet count along with elevated WBC count, lactate dehydrogenase, indirect bilirubin, and total bilirubin.
Microscopic image showing schistocytes (black arrow) (100x).
Investigation performed post emergency laparotomy.
| Investigations | Findings | Normal ranges |
|---|
| Haemoglobin (Hb) (g/dL) | 7.2 | 12.0-15.5 (females) |
| White blood cells (cells/μL) | 14000 | 4,000-11,000 |
| Platelets (/μL) | 65000 | 150,000-450,000 |
| Urea (mg/dL) | 30 | 7-20 |
| Creatinine (mg/dL) | 1.2 | 0.6-1.2 (females) |
| Indirect bilirubin (mg/dL) | 1.5 | 0.1-1.0 |
| Total bilirubin (mg/dL) | 2.5 | 0.1-1.2 |
| Lactate Dehydrogenase (LDH) (U/L) | 1000 | 140-280 |
| C-Reactive Protein (CRP) (mg/L) | 0.79 | <3.0 |
POD 1: Despite stable vitals, the rapid drop in platelet count from 65,000/μL to 26,000/μL, along with a significant decline in Hb to 5.7 mg/dL, raised suspicion of an underlying platelet disorder. One unit of packed red blood cells was transfused. Her serum was sent for evaluation, particularly to detect autoantibodies that could destroy platelets or red blood cells; the results were positive for Anti-Rho antibodies. Other findings are presented in [Table/Fig-6], with a positive Anti-nuclear Antibody titer of 1/320 and elevated Anti-dsDNA antibodies of 39.24 IU/mL, suggesting an underlying autoimmune condition.
Anti-nuclear antibodies profile.
| Investigation | Findings | Normal values |
|---|
| Anti-nuclear antibodies | 1/320 Titer (Positive) | Often <1:40 titer |
| Anti-ds DNA (IU/mL) | 39.24 (Positive) | <30 |
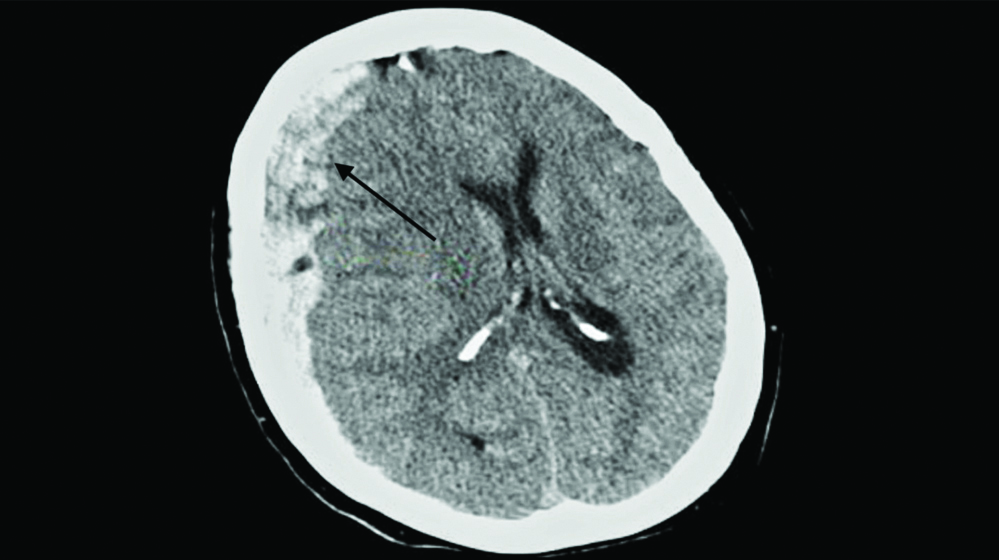
POD 2: Her condition worsened, with platelet counts dropping to 7,000/μL, resulting in massive haematuria and Hb dropping to 5.9 mg/dL. A complete urine examination confirmed gross haematuria. Two units of packed red blood cells were transfused for stabilisation. Later that day, she abruptly developed severe neurological symptoms, including an intense headache, confusion, altered mental status, paresthesia and hemiparesis in the left upper and lower limbs. An urgent Computed Tomography (CT) brain scan showed subdural haemorrhage with a midline shift [Table/Fig-7], signifying intracranial bleeding and increased intracranial pressure. She was immediately started on i.v. mannitol and was thoroughly monitored. As the patient was unfit for surgery, no surgical intervention was performed.
CT image showing right subdural haemorrhage in black arrow.
The findings mentioned above, along with the presence of malar erythema, raised suspicion of SLE, which was confirmed through immunological tests including anti-nuclear antibody, anti-dsDNA, and anti-Rho antibodies.
From POD 3 to 5: The patient improved clinically, with platelet counts increasing from 40,000/μL on POD3 to 58,000/μL on POD5, and Hb levels between 7.8-8.6 mg/dL, indicating partial recovery. However, her neurological deficits persisted, including altered cognition and weakness, despite the stabilisation of haematological parameters. To treat and assess her potential recovery, continuous medical opinions were sought. Given multisystem involvement, a clinical diagnosis of SLE was made, and i.v. methylprednisolone was initiated as part of her therapy. Renal and neurological status was closely monitored by the multidisciplinary team.
On POD 6: The patient experienced generalised tonic-clonic seizures, indicating further neurological deterioration. Her platelet count dropped to 35,000/μL, and her Hb was 7.5 mg/dL. Intravenous (i.v.) valproic acid and four units of SDPs were administered. The neurological deficits, including seizures, coupled with her persistently low platelet count and clinical findings, led to suspicion of TTP. The PLASMIC score was evaluated and found to be 5, indicating a high probability of TTP. Based on the PLASMIC score, ADAMTS13 activity was evaluated, which was 67%. The patient also tested positive for anti-ADAMTS13 antibodies. Further serological evaluations for immune-mediated haemolysis and complement activation are depicted in [Table/Fig-8], showing a positive Direct Coombs test, specifically for C3d. This led to the diagnosis of TTP. Targeted TTP management was initiated, including corticosteroids and immunosuppressive agents to reduce autoimmune activity, and further plans were made for plasma exchange therapy to achieve a rapid therapeutic response by removing autoantibodies.
Additional serological evaluations for immune mediated haemolysis and complement activation.
| Investigations | Findings | Normal value |
|---|
| Direct coombs test | +1 | Negative |
| Indirect coombs test | Negative | Negative |
| Auto control (4°C) | + (Positive) | 0 (Negative) |
| C3d (Monospecific) | + (Positive) | Negative |
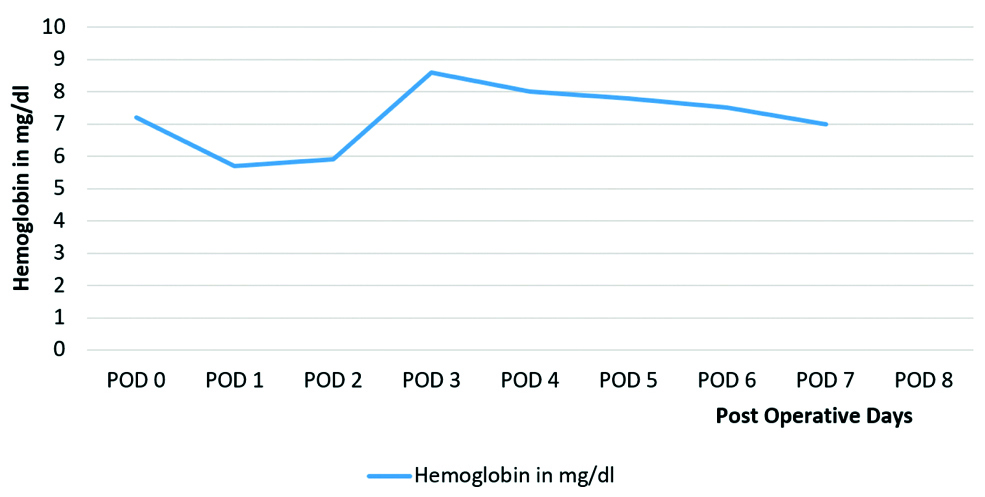
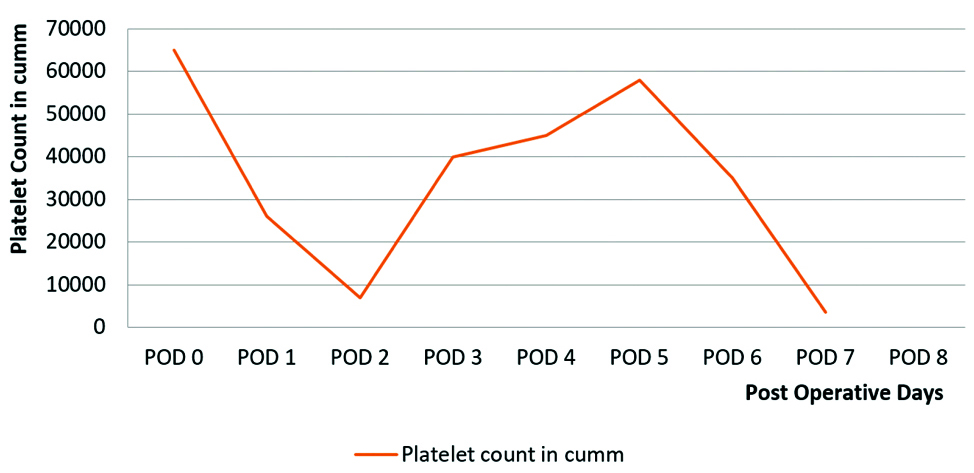
On POD 7: The patient’s condition worsened with cardiovascular collapse, and the platelet count dropped to 8,000/μL, requiring an immediate platelet transfusion. This resulted in a temporary improvement to 15,000/μL, but it dropped again to 3,500/μL within 24 hours, and her Hb was 7 mg/dL. Changes in the patient’s Hb level from admission are demonstrated in [Table/Fig-9]. Haemodynamic instability and extreme fluctuations in platelet counts, depicted in [Table/Fig-10], rendered her unfit for plasma exchange. Further serological and renal function tests were conducted; key findings are included in [Table/Fig-11]. Aggressive management with valproic acid to prevent seizures and platelet transfusions continued with careful monitoring over the next 18 hours.
Graphical depiction of the changes in Haemoglobin (Hb) levels during admission.
Graphical depiction of the change in Platelet count during admission.
Renal function and complete blood picture on postoperative day 7.
| Investigation | Findings | Normal range |
|---|
| Creatinine (mg/dL) | 2.1 | 0.6-1.2 (females) |
Blood Urea Nitrogen (BUN)
(mg/dL) | 35 | 7-20 |
| Haemoglobin (Hb) (g/dL) | 7 | 12.0-15.5 (females) |
| White blood cells count (cells/μL) | 12,000 | 4,000-11,000 |
| Platelets count (/μL) | 8,000,
After 6 hours of platelets transfusion 15,000 | 150,000-450,000 |
On POD 8: Despite aggressive interventions, she deteriorated further and experienced another cardiovascular collapse. Resuscitation efforts were unsuccessful, and she succumbed to her illness. This unfortunate outcome highlights the severe course of TTP with multiorgan involvement, particularly when the disease becomes refractory and unresponsive to standard interventions.
Discussion
The SLE is an autoimmune condition that mainly affects women of childbearing age. It can rarely co-exist with TTP, sharing common mechanisms like autoantibody production and complement activation, which is approximately seen in 0.5% of SLE patients [1,2]. The overlapping clinical features, such as thrombocytopenia, haemolytic anaemia and renal dysfunction, complicate the diagnosis and treatment, increasing the risk of morbidity and mortality. Therefore, a thorough understanding of the pathophysiological interactions between TTP and SLE is essential for risk assessment and guiding therapeutic approaches, highlighting the need for ongoing research to improve patient care and develop more effective long-term treatment options.
The pathogenesis of SLE-associated TTP focuses on the deficiency or inhibition of ADAMTS13, a protease that cleaves large vWF multimers, leading to the accumulation of vWF, platelet aggregation, and microvascular thrombosis [3]. Microvascular injury, which is also caused by elevated cytokines including Interleukin-6 (IL-6) and Tumour Necrosis Factor alpha (TNF-α), leads to consequences like MAHA, immune-mediated platelet destruction, tissue ischaemia, and organ damage, specifically to the brain and kidneys. MAHA is observed as schistocytes on a blood smear, resulting from fragmented red blood cells in the obstructed microvasculature [4,5]. This case emphasises how autoimmune TTP, particularly when associated with SLE, can present in a fulminant manner with rapid multiorgan involvement [6].
This patient was diagnosed with SLE due to physical examination findings such as malar rash, petechiae, purpura, decreased platelet count and positive tests for anti-nuclear antibody, anti-dsDNA, and anti-Rho antibodies. The mildly raised serum creatinine, Blood Urea Nitrogen (BUN), and haematuria can be explained by renal involvement in SLE-lupus nephritis [7,8]. Neurological manifestations like confusion, paresthesia, and ultimately seizures can be attributed to either Neuropsychiatric Lupus (NPSLE) or TTP-related neurological involvement. Clinically, TTP presents with a pentad of symptoms that includes fever, renal and neurological abnormalities, haemolytic anaemia, and thrombocytopenia. The overlap of these clinical features between TTP and SLE complicated the diagnosis and management of this patient [9]. Haematological malignancies, such as Chronic Myeloid Leukaemia (CML), should also be included in the differential diagnosis, as they could potentially present with similar haematological and systemic features [10].
The PLASMIC score, which generally ranges from 0 to 7 and includes assessments of platelet count, evidence of haemolysis, and neurological findings, was noted as five, indicating a high probability of TTP [11]. A deficiency of ADAMTS13, due to auto-antibodies induced against ADAMTS13, can lead to TTP. An ADAMTS13 activity level of <5 and positive anti-ADAMTS13 antibodies were found in this patient, confirming the diagnosis of autoimmune TTP. Additionally, a positive direct Coombs test for C3d and low platelet counts also support an immune-mediated pathogenesis, where haematological abnormalities are likely driven by complement activation.
The standard therapy for SLE flares, which is intravenous methylprednisolone, was administered to the patient and also helped to stabilise platelet counts. However, the patient’s unresponsiveness to platelet transfusions and the rapid decline in platelet count signaled refractory TTP. Plasma exchange, which involves the removal of antibodies, is the cornerstone treatment for a rapid therapeutic response in TTP. It was initially planned, but the patient’s cardiovascular collapse made it unfeasible. Extreme fluctuations in platelet counts, combined with worsening renal impairment (evident from laboratory findings showing rising creatinine and BUN) and neurological decline, reflected the systemic severity of the refractory condition [12]. A multidisciplinary team is essential to monitor changes in platelet count and alterations in neurological and renal functions. Despite all these interventions, the patient’s condition deteriorated, leading to multiorgan failure and death.
This case underscores the importance of differentiating between SLE flare and TTP, as it results in a complicated course, as seen in this case. The superimposed thrombotic and autoimmune aetiologies resulted in rapid symptomatic worsening, complicating prompt and accurate diagnosis and early initiation of appropriate therapy for a favourable therapeutic response [13].
Conclusion(s)
This case illustrates the severity and fatal outcomes of overlapping autoimmune diseases, as seen in this young female with TTP and SLE. Neurological and renal manifestations, along with thrombocytopenia in a patient previously diagnosed with SLE, can mask the diagnosis of TTP. These cases highlight the importance of early diagnosis and prompt treatment. They also demand high-standard diagnostic protocols and markers, as well as further intensive research on various treatment strategies and the involvement of multidisciplinary teams in managing severe, refractory, and overlapping autoimmune conditions with multiorgan involvement.
[1]. George JN, Terrell DR, Swisher KK, Vesely SK, Lessons learned from the Oklahoma thrombotic thrombocytopenic purpura-hemolytic uremic syndrome registryJ Clin Apher 2008 23(4):129-37.10.1002/jca.2016918618590 [Google Scholar] [CrossRef] [PubMed]
[2]. Tsai HM, Pathophysiology of thrombotic thrombocytopenic purpuraInt J Hematol 2010 91(1):01-19.10.1007/s12185-009-0476-120058209PMC3159000 [Google Scholar] [CrossRef] [PubMed]
[3]. Fakhouri F, Vernant JP, Veyradier A, Wolf M, Kaplanski G, Binaut R, Efficiency of curative and prophylactic treatment with rituximab in ADAMTS13-deficient thrombotic thrombocytopenic purpura: A study of 11 casesBlood 2005 106(6):1932-37.Epub 2005 Jun 210.1182/blood-2005-03-084815933059 [Google Scholar] [CrossRef] [PubMed]
[4]. George JN, Clinical practice. Thrombotic thrombocytopenic purpuraN Engl J Med 2006 354(18):1927-35.10.1056/NEJMcp05302416672704 [Google Scholar] [CrossRef] [PubMed]
[5]. Sadler JE, Pathophysiology of thrombotic thrombocytopenic purpuraBlood 2017 130(10):1181-88.Epub 2017 Aug 210.1182/blood-2017-04-63643128768626PMC5606001 [Google Scholar] [CrossRef] [PubMed]
[6]. Yue C, Su J, Fan X, Song L, Jiang W, Xia J, Immune-mediated thrombotic thrombocytopenic purpura in patients with and without systemic lupus erythematosus: A retrospective studyOrphanet J Rare Dis 2020 15(1):22510.1186/s13023-020-01510-932859237PMC7456051 [Google Scholar] [CrossRef] [PubMed]
[7]. Hanly JG, O’Keeffe AG, Su L, Urowitz MB, Romero-Diaz J, Gordon C, The frequency and outcome of lupus nephritis: Results from an international inception cohort studyRheumatology 2016 55(2):252-62.10.1093/rheumatology/kev31126342222PMC4939728 [Google Scholar] [CrossRef] [PubMed]
[8]. Ponticelli C, Moroni G, Flares in lupus nephritis: Incidence, impact on renal survival and managementLupus 1998 7(9):635-38.10.1191/0961203986789207769884102 [Google Scholar] [CrossRef] [PubMed]
[9]. Rutherford C, Immune thrombocytopenic purpura: An updateInternal Medicine Grand Rounds 2010 Dallas (TX)UT Southwestern Medical CenterAvailable from: https://utswmed-ir.tdl.org/items/80d2bbb4-11b3-40bb-a0ac-206c45d887a7 [Google Scholar]
[10]. Kanani J, Sheikh MI, An autopsy presentation of spontaneous splenic rupture in chronic myeloid leukemia: A rare case reportJ Med Surg Public Health 2024 2024:10011810.1016/j.glmedi.2024.100118 [Google Scholar] [CrossRef]
[11]. Paydary K, Banwell E, Tong J, Chen Y, Cuker A, Diagnostic accuracy of the PLASMIC score in patients with suspected thrombotic thrombocytopenic purpura: A systematic review and meta-analysisTransfusion 2020 60(9):2047-57.10.1111/trf.1595432757237 [Google Scholar] [CrossRef] [PubMed]
[12]. Kojouri K, Vesely SK, George JN, Quinine-associated thrombotic thrombocytopenic purpura-hemolytic uremic syndrome: Frequency, clinical features, and long-term outcomesAnn Intern Med 2001 135(12):1047-51.10.7326/0003-4819-135-12-200112180-0000811747383 [Google Scholar] [CrossRef] [PubMed]
[13]. Hamasaki K, Mimura T, Kanda H, Kubo K, Setoguchi K, Satoh T, Systemic lupus erythematosus and thrombotic thrombocytopenic purpura: A case report and literature reviewClin Rheumatol 2003 22(4-5):355-58.10.1007/s10067-003-0742-114579168 [Google Scholar] [CrossRef] [PubMed]