Introduction
Dyke-Davidoff-Masson Syndrome (DDMS) was initially reported in 1933 by radiologist Cornelius G. Dyke (1900-1943), neurosurgeon Leo M. Davidoff (1898-1975) and neurosurgeon Clement M. Masson, from the X-ray and surgical Departments of the Neurological Institute of New York [1]. DDMS is an uncommon neurological disorder characterised by a range of symptoms. Despite its rarity, the exact prevalence of DDMS remains uncertain due to limited research, inconsistent diagnostic criteria and similarities with other conditions. Available estimates suggest that DDMS affects approximately one in 100,000 to one in 500,000 newborns and accounts for 0.4% to 1.4% of cerebral palsy cases. The condition exhibits a male predominance, with a male-to-female ratio of 2:1.
Case Series
Case 1
A 22-year-old female patient presented with repeated episodes of generalised seizure disorder and left-sided hemiparesis for 15 years. She reported 10-15 brief seizure episodes lasting 10-30 seconds per month, with moderate intensity (Glasgow Coma Scale score 3-5). She was unable to walk or speak normally at the time of examination. Her parents provided a history, stating that the age of onset was in early childhood. She had developmental delays and was intellectually disabled. She was taken to a local practitioner by her parents (the treatment record was unavailable).
Her birth history was normal and there was no history of trauma. The patient was dependent on her parents for daily activities and her siblings were normal. On examination, she exhibited poor cognitive function and was undernourished. Additional observations included brisk deep tendon reflexes and power zero in the left upper and lower limbs. Examination of other systems was grossly normal.
For patients diagnosed with epilepsy, the Medicine department of the hospital follows a routine protocol that includes biochemical and pathological tests to inform treatment and management plans. Specific tests may include blood tests (e.g., serum electrolytes, liver function tests, renal function tests, complete blood count).
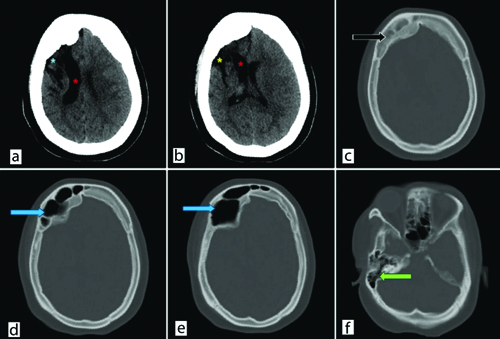
The results of blood investigations were unremarkable and within the normal range. CT imaging of the same patient showed gliotic changes with hemiatrophy of the right cerebral hemisphere and ipsilateral dilatation of the right lateral ventricle. Bony calvarial thickening was noted and hyperpneumatisation of the ipsilateral frontal sinus was observed [Table/Fig-1a-f]. These radiological and clinical indicators led to the diagnosis of DDMS.
a and b) Non Contrast Computed Tomography (NCCT) head shows right-sided gliotic and encephalomalacia changes involving frontoparietal lobe (blue asterisk) with ex-vacuo dilatation of right lateral ventricle involving frontal horn and body (red asterisks) with resultant right hemicerebral atrophy. Prominent right sylvian fissure (yellow asterisk); c) Bone window shows calvaria thickening (black arrow); d and e) Hyperpneumatisation of right frontal sinus (blue arrows); f) Hyperpneumatisation of right mastoid air cells (green arrow).
The patient’s treatment regimen includes anticonvulsants (phenytoin 15 mg/kg) and muscle relaxants (baclofen) to control seizures and muscle spasms. A multidisciplinary rehabilitation plan has been initiated, encompassing physical therapy for mobility and strength, occupational therapy for daily living skills, speech therapy for communication, cognitive training for intellectual disability and psychiatric support for emotional well-being. The patient has been educated on the benefits and potential side effects of medication and is scheduled for regular follow-up appointments in the medicine Outpatient Department (OPD). With appropriate seizure crisis management and a sufficient dosage of antiepileptic medication, the patient is presently stable.
Case 2
A 15-year-old female patient presented with abnormal jerky movements of the right upper and lower limbs for the past 10 years and was referred for a CT scan. She was a known case of seizure disorder, with episodes occurring 10-15 times per month, lasting 10-30 seconds, with mild intensity and was on phenytoin medication for the past 10 years. This patient has a history of hospital admission for one month following birth due to perinatal complications. Unfortunately, the patient’s medical records from the previous hospitalisation were unavailable. She was well-oriented to time, place and person but exhibited right-sided weakness. Lower limb and upper limb power were 3/5 and 2/5, respectively. Her cognitive behaviour tended to be normal and her Intelligence Quotient (IQ) was average. The patient was undernourished.
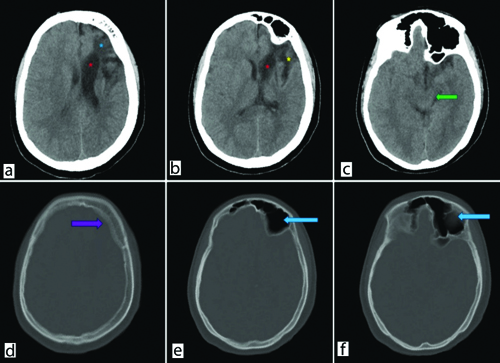
On Computed Tomography (CT) scan [Table/Fig-2a-f], gliotic and encephalomalacia changes were observed in the left frontoparietal region with dilatation of the left lateral ventricle. Left-sided cerebral hemiatrophy, left-sided midbrain atrophy likely due to Wallerian degeneration and cerebellar atrophy were noted. Hyperpneumatisation of the left frontal sinus and bilateral mastoid air cells, along with diffuse calvarial thickening, were also observed. After carefully ruling out various neurological diseases that can present with similar symptoms, especially Sturge-Weber syndrome and Rasmussen encephalitis, our patient was diagnosed with DDMS.
a and b) Non contrast Computed Tomography (NCCT) Brain axial images demonstrate left sided gliotic changes involving frontal lobe (blue asterisk) with ex-vacuo dilatation of left lateral ventricle (red asterisks). Prominent left sylvian fissure (yellow asterisk); c) Left-sided midbrain atrophy (green arrow); d) Bone window image shows bony calvaria thickening (violet arrow) along left side of frontal bone; e and f) Hyper pneumatisation of left frontal sinus (blue arrows).
The patient responded well to antiseizure medication (phenytoin mg/kg). She was also advised to follow a comprehensive rehabilitation program incorporating physical, occupational, speech and cognitive therapies, along with psychiatric support.
Case 3
A 21-year-old male presented with a head injury after falling from his bed. His past medical history included right-sided hemiparesis, which began in childhood and had persisted for 15 years. The patient had a non specific birth history with complaints of hemiparesis since early life. He was not well oriented to time, place, or person. As per standard protocol, the patient underwent a CT scan of the brain for the head injury.
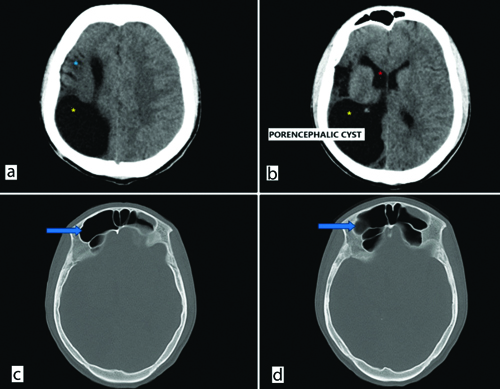
On Computed Tomography (CT) scan [Table/Fig-3a-d], gliotic changes in the right parietal lobe were observed, along with right-sided cerebral atrophy and ex-vacuo dilatation of the frontal horn of the right lateral ventricle. A large porencephalic cyst was present, involving the right parieto-temporo-occipital region and communicating with the right lateral ventricle. Hyperpneumatisation of the bilateral frontal sinuses was noted, more on the right side, with marginal thickening of the bony calvaria. The co-existence of DDMS and a porencephalic cyst sets this patient’s case apart. As there were no cutaneous manifestations or intracranial lipoma, Fishman syndrome was ruled out [2].
a and b) Non contrast Computed Tomography (NCCT) brain axial image demonstrates gliotic volume loss in right parietal region (blue asterisk), dilatation of frontal horn of right lateral ventricle (red asterisk) and a large porencephalic cyst (yellow asterisk) communicating with right lateral ventricle (internal type); c and d) Hyper pneumatisation of bilateral frontal sinus more on right-side (blue arrows). Marginal thickening of bony calvaria on right-side in frontal region.
The patient’s treatment plan is symptom-driven, emphasising convulsion control and comprehensive rehabilitation (physical and mental). Following medication counselling, covering benefits and potential side effects, regular follow-up appointments have been arranged in the Medicine OPD.
Discussion
The DDMS is a combination of brain parenchymal and calvarial changes. The literature describes three hallmark features of DDMS: cerebral hemiatrophy with ex-vacuo dilation of the lateral ventricle, ipsilateral thickening of the skull’s diploic space and ipsilateral hyperpneumatisation of the frontal sinuses [1]. Rondão MBA et al., show that the average age of patients was 19.44 years, with males showing a slightly higher prevalence [3]. DDMS is primarily categorised into two types: acquired and congenital. Any damage to the brain parenchyma occurring during pregnancy (in utero) is classified as congenital, whereas brain injuries resulting from infections, trauma, subependymal bleeds and perinatal hypoxic injuries sustained after birth are classified as acquired.
The underlying etiology of this syndrome is hypothesised to involve any premature brain insult, which leads to gliosis, hemi-atrophy of the brain and compensatory overgrowth of the overlying bony calvaria, particularly in the frontal region. This is attributed to delayed maturation and pneumatisation of the frontal sinus postnatally. Prolonged ischaemia at any point from foetal development to 3-4 years of age can cause a decrease in brain growth factors, ultimately leading to hemi-atrophy [4,5]. Early brain insults are associated with progressively more severe changes in brain parenchyma.
Patients mainly complain of seizure disorders and variable degrees of cognitive dysfunction [6,7]. Patients exhibit variable degrees of muscle weakness [7]. Similar complaints were observed in the patient. A case report indicates that patients can be asymptomatic despite having positive anatomical findings [8]. This suggests that the spectrum of patients with the disease is much broader, as the domain of asymptomatic patients often goes undiagnosed.
Sharma S et al., presented a case of a 12-year-old female with DDMS. According to their findings, CT is the diagnostic modality of choice in DDMS [9]. In resource-limited settings where Magnetic Resonance Imaging (MRI) is unavailable, CT plays a key role in identifying patients with DDMS. It is a cheaper and quicker modality available in most tertiary care centers. Typical findings such as hemiatrophy and bony changes can be observed on CT.
Durcan R et al., reported MRI findings of DDMS in a 50-year-old male patient that revealed similar alterations [10]. Our two female patients presenting with seizure disorders showed findings on Non Contrast Computed Tomography (NCCT) head scans that are consistent with DDMS. A rare and intriguing association between DDMS and a porencephalic cyst was observed in our third case. Given that porencephalic cysts and DDMS share a common aetiopathogenesis, specifically early brain parenchymal insult during development, it is plausible to observe both conditions concurrently, as seen in the third patient. Although this association is not well-established in the existing literature, it should not be dismissed as a potential correlation.
Anti-seizure medication is the mainstay of treatment, which is coupled with physiotherapy to reduce seizures [6]. While monotherapy may not be very effective, close follow-up, dose adjustments, a combination of drugs and supportive care are necessary to decrease the morbidity of the patient.
The aforementioned patients were treated conservatively following the diagnosis of DDMS. These individuals received phenytoin, rehabilitative therapy and lifestyle modifications.
Differential diagnosis:
Sturge-Weber syndrome: Bony calvaria changes may be similar to DDMS, but cutaneous angiomas are present. A wine stain easily distinguishes it from DDMS diagnosis [11].
Fibrous dysplasia of the skull: Thickening of the bony calvaria with ground glass opacity is present however, the underlying brain parenchyma is normal. There is no any evidence of brain injury or parenchymal insult [12].
Rasmussen’s encephalitis: In these patients, brain findings are similar to those in DDMS, but the calvaria are normal [13].
Hemimegaloencephaly: In these patients, the contralateral cerebral hemisphere appears atrophied while the other half becomes hypertrophied, leading to ventricular enlargement [14].
Parry-Romberg syndrome: Patients primarily present with progressive hemifacial palsy and atrophy. Other features may include dystrophic calcifications, porencephaly and multiple cavernomata [15].
Conclusion(s)
The DDMS should be considered in the differential diagnosis of unresponsive seizure disorders accompanied by hemiparesis. A thorough clinical history, along with signs, symptoms and imaging features, is essential for an accurate diagnosis. In low-resource settings, CT is a vital diagnostic tool. NCCT of the brain readily detects changes in brain parenchyma and the bony calvaria. Following the diagnosis, appropriate counselling and treatment can be tailored to the patient’s needs.
[1]. Lanska DJ, Cruveilhier’s unrecognized case (c1831) of Dyke-Davidoff-Masson SyndromeEur Neurol 2021 84(4):300-06.Epub 2021 May 710.1159/00051580833965957 [Google Scholar] [CrossRef] [PubMed]
[2]. Chandravanshi SL, Encephalocraniocutaneous lipomatosis: A case report and review of the literatureIndian J Ophthalmol 2014 62(5):622-27.10.4103/0301-4738.13352124881613PMC4065518 [Google Scholar] [CrossRef] [PubMed]
[3]. Rondão MBA, Hsu BR, Centeno RS, de Aguiar PH, Dyke-Davidoff-Masson syndrome: Main clinical and radiological findings-Systematic literature reviewSeizure 2023 110:58-68.10.1016/j.seizure.2023.04.02037327751 [Google Scholar] [CrossRef] [PubMed]
[4]. Adebayo PB, Bakare A, Bello MM, Olaewe OD, Wahab KW, Dyke-Davidoff Masson syndrome in a NigerianEpilepsy Behav Case Rep 2017 7:10-12.10.1016/j.ebcr.2016.09.00328053860PMC5199153 [Google Scholar] [CrossRef] [PubMed]
[5]. Roy U, Panwar A, Mukherjee A, Biswas D, Adult presentation of Dyke-Davidoff-Masson syndrome: A case reportCase Rep Neurol 2016 8(1):20-26.10.1159/00044352126933427 [Google Scholar] [CrossRef] [PubMed]
[6]. Al-Attas AA, Alwazna EO, Presentation of Dyke-Davidoff-Masson syndrome in adult maleNeurosciences (Riyadh) 2023 28(2):143-47.10.17712/nsj.2023.2.2022008437045464PMC10155480 [Google Scholar] [CrossRef] [PubMed]
[7]. Diestro JD, Dorotan MK, Camacho AC, Perez-Gosiengfiao KT, Cabral-Lim LI, Clinical spectrum of Dyke-Davidoff-Masson syndrome in the adult: An atypical presentation and review of literatureCase Rep 2018 2018:bcr-2018.10.1136/bcr-2018-22417029973410PMC6040550 [Google Scholar] [CrossRef] [PubMed]
[8]. Lammle M, Gilbert C, Nagel JE, Hagan EA, Dyke-Davidoff-Masson syndrome: Imaging diagnosis in an asymptomatic adultRadiol Case Rep 2022 17(11):4328-31.10.1016/j.radcr.2022.08.06036132061PMC9483625 [Google Scholar] [CrossRef] [PubMed]
[9]. Sharma S, Goyal D, Negi A, Sood RG, Jhobta A, Surya M, Dyke-davidoff masson syndromeIndian J Radiol Imaging 2006 16(02):165-66.10.4103/0971-3026.29077 [Google Scholar] [CrossRef]
[10]. Durcan R, Smyth S, Bolster F, Teaching neuroimages: Dyke-Davidoff-Masson syndromeNeurology 2018 90(23):e2097-98.10.1212/WNL.000000000000564029866944 [Google Scholar] [CrossRef] [PubMed]
[11]. Poliner A, Fernandez Faith E, Blieden L, Kelly KM, Metry D, Port-wine birthmarks: Update on diagnosis, risk assessment for Sturge-Weber syndrome, and managementPediatr Rev 2022 43(9):507-16.10.1542/pir.2021-00543736045161 [Google Scholar] [CrossRef] [PubMed]
[12]. DiCaprio MR, Enneking WF, Fibrous dysplasia: Pathophysiology, evaluation, and treatmentJBJS 2005 87(8):1848-64.10.2106/00004623-200508000-00028 [Google Scholar] [CrossRef]
[13]. Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, Rasmussen’s encephalitis: Clinical features, pathobiology, and treatment advancesLancet Neurol 2014 13(2):195-205.10.1016/S1474-4422(13)70260-624457189 [Google Scholar] [CrossRef] [PubMed]
[14]. Sugiyama S, Fujii M, Nomura S, Yamashita T, Ito H, Hayashi T, Hemimegaloencephaly with periventricular heterotopia- Case reportNeurologia Med Chir (Tokyo) 1994 34(8):561-64.10.2176/nmc.34.5617526246 [Google Scholar] [CrossRef] [PubMed]
[15]. Knights H, Minas E, Khan F, Shaw L, Al Obaidi M, Mankad K, Magnetic resonance imaging findings in children with Parry-Romberg syndrome and en coup de sabrePediatr Rheumatol Online J 2021 19(1):4210.1186/s12969-021-00512-633757522PMC7986399 [Google Scholar] [CrossRef] [PubMed]