Guillain-Barré syndrome (GBS) is the most common cause of acute flaccid paralysis in children. The clinical variants include Acute Inflammatory Demyelinating Polyradiculoneuropathy (AIDP), Acute Motor Sensory Axonal Neuropathy (AMSAN), Acute Motor Axonal Neuropathy (AMAN) and Miller-Fisher syndrome. Cranial nerve involvement can occur in approximately 50% of patients with GBS, but it is rarely reported in the AMAN variant. Ptosis as a presenting symptom is extremely rare in the AMAN variant. Autonomic dysfunction is also rarely reported in the AMAN variant. In the present case series, authors hereby, report five children (3 males, 2 females, aged 4-11 years) diagnosed with the AMAN variant of GBS, who presented with early onset of ptosis and rapid progression of flaccid weakness. Autonomic dysfunction, in the form of tachycardia, hypertension and excessive diaphoresis, was a prominent feature in four of the cases. The average time to reach the nadir of weakness was 4.2 days and the average duration of hospital stay was 58 days. The average GBS disability score at discharge was four. Nerve conduction studies were suggestive of the AMAN variant of GBS in all five cases. All children were treated with Intravenous Immunoglobulin (IVIg) and all required mechanical ventilation, with an average duration of 35.8 days. All five cases presented in a short period of three months, from November 2022 to February 2023.
Introduction
The GBS is the most common cause of acute flaccid paralysis in children [1]. GBS can present in several variants: a classic demyelinating form- AIDP, less common types such as AMSAN, AMAN, Miller–Fisher syndrome and other rarer forms [2]. Cranial nerve involvement can occur in approximately 50% of patients with GBS, but it is rarely reported in the AMAN variant [3]. Ocular involvement in GBS can occur in 10% of cases, although ptosis is rare [3,4]. To the best of authors knowledge, there are no reported cases of the AMAN variant of GBS presenting with ptosis without ophthalmoplegia in children. Autonomic dysfunction is also rarely reported in AMAN [5]. In the present case series, authors present an unusual clustering of five cases of AMAN in a short period, with early onset of ptosis and severe autonomic symptoms. The clinical and laboratory characteristics are summarised in [Table/Fig-1].
Clinical and laboratory characteristics.
| Parameters |
|---|
| Age in years | 9 | 6 | 11 | 4 | 5 |
|---|
| Gender | Male | Female | Male | Male | Female |
| Preceding infection/immunisation | Upper respiratory infection | Diarrhoea | Upper respiratory infection | Upper respiratory infectionDPT+MMR vaccine one month prior | Upper respiratory infection |
| Cranial nerve involvement | Ptosis, facial and bulbar palsy | Ptosis, facial and bulbar palsy | Ptosis, facial and bulbar palsy | Ptosis, facial and bulbar palsy | Ptosis, facial and bulbar palsy |
| Autonomic symptomsTachycardia, hypertensionIncreased sweating | Yes | Yes | Yes | No | Yes |
| Duration of ventilation | 23 days | 60 days | 75 days | 6 days | 15 days |
| Time to reach nadir | 3 days | 5 days | 24 hours | 5 days | 7 days |
| Time to start Intravenous Immunoglobulin (IVIg) | 24 hours | 48 hours | 24 hours | 5 days | 3 days |
| Duration of hospital stay | 60 days | 83 days | 97days | 15 days | 35 days |
| Nerve conduction study(upper and lower limbs)CMAP*SNAP† | AbsentNormal | AbsentNormal | AbsentNormal | AbsentNormal | AbsentNormal |
| CSF protein | 255 mg/dL | 275 mg/dL | 398mg/dL | 10 mg/dL | 108 mg/dL |
| CSF cells | <5 cells | <5 cells | <5 cells | <5 cells | 10 cells |
| SerumAntiGQ1b, AntiGD1a AntiGD1b, AntiGM1, AntiGM2, Anti GM3antibodies | Negative | Negative | Negative | Not done | Negative |
*CMAP: Compound muscle action potential; †SNAP: Sensory nerve action potential
Case 1
A nine-year-old boy, with a history of mild upper respiratory infection two weeks prior, presented with an acute onset of weakness in the lower limbs, which rapidly progressed over the next 12 hours to involve the upper limbs. At admission, he exhibited normal higher functions, bilateral ptosis without ophthalmoplegia [Table/Fig-2] and bilateral facial and bulbar palsy. There was hypotonia and areflexia, with power graded at 3/5 in all four limbs, which progressed to 0/5 power within two days, necessitating mechanical ventilation.
Ptosis in AMAN Variant of Guillain-Barré syndrome (GBS) in case 1.
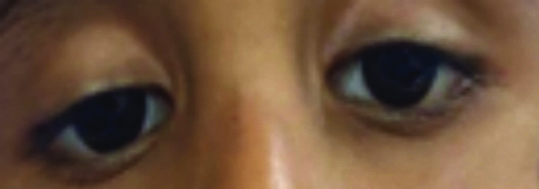
The differential diagnoses considered at this point included GBS, specifically the Miller-Fisher variant (MFS) and Myasthenia Gravis (MG). Complete blood count, C-reactive protein, serum electrolytes, as well as renal and liver function tests were normal. Nerve conduction studies revealed an absence of Compound Motor Action Potential (CMAP) with preserved Sensory Nerve Action Potential (SNAP) in both upper and lower limbs, suggestive of the AMAN variant of GBS. Cerebrospinal Fluid (CSF) analysis showed albumin-cytological dissociation. Antibodies against GQ1b, GD1a, GD1b, GM1, GM2 and GM3 were negative. Magnetic resonance imaging (MRI) of the brain and spine screening appeared normal.
He was treated with IVIg at a dosage of 2 g/kg over five days, along with other supportive measures. He also exhibited features of autonomic dysfunction, including hypertension, tachycardia, increased diaphoresis and frequent ventricular ectopics by the end of the first week, which lasted for three weeks. He remained on the ventilator for 23 days. Ptosis began to improve at the end of the second week of illness and completely resolved by week four. The GBS disability score at discharge on day 60 was 4. He had a good recovery by the end of one year, with a minor foot drop, resulting in a GBS disability score of 1.
Case 2
A six-year-old girl presented with weakness in both lower limbs and drooping of the eyelids, which had lasted for one day. There was a history of loose stools five days prior to the onset of weakness. On examination, she exhibited normal higher functions, bilateral ptosis (left more than right) without ophthalmoplegia [Table/Fig-3], bilateral facial and bulbar weakness, hypotonia, areflexia and weakness in both lower limbs (2/5 power in all muscle groups), which progressed to the upper limbs, trunk and neck muscles over the next 24 hours. Complete blood count, C-reactive protein, serum electrolytes and renal and liver function tests were normal. Nerve conduction studies revealed the absence of CMAP with preserved SNAP, suggestive of the AMAN variant of GBS. CSF analysis revealed albumino-cytological dissociation. Antibodies against GQ1b, GD1a, GD1b, GM1, GM2 and GM3 were negative. She was treated with intravenous IVIg at a dose of 2 g/kg over five days. By day three of hospitalisation, her weakness progressed to 0/5 power in all limbs and she required mechanical ventilation, which continued for 60 days. As there was no significant improvement in weakness after two weeks, a repeat course of IVIg was administered. She also exhibited features of autonomic dysfunction, including hypertension, tachycardia, bradycardia and excessive sweating. Ptosis began to improve at the start of the third week. The GBS disability score at discharge on day 83 was four. She experienced significant recovery at the end of one year, with minor foot drop and a GBS disability score of 2.
Ptosis in AMAN Variant of Guillain-Barré syndrome (GBS) in case 2.

Case 3
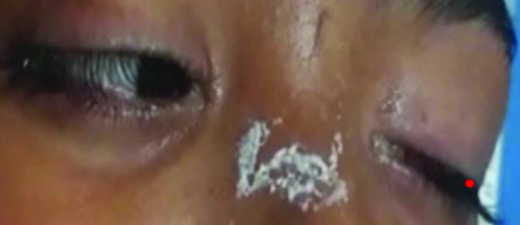
An 11-year-old boy with a history suggestive of an upper respiratory infection one week prior presented with weakness in both lower limbs, which rapidly progressed to involve all limbs, requiring mechanical ventilation within 16 hours of presentation. On examination, his higher mental functions were normal, but he exhibited bilateral lower motor neuron facial palsy and left-sided ptosis without ophthalmoplegia [Table/Fig-4]. The ptosis later became bilateral. Muscle power was 0/5 in all limbs, accompanied by hypotonia and hyporeflexia. He also showed features of autonomic dysfunction, including tachycardia, bradycardia, hypertension and excessive sweating. Complete blood count, C-reactive protein, serum electrolytes, renal and liver function tests were all normal. Nerve conduction studies revealed the absence of CMAP with preserved SNAP, suggestive of the AMAN variant of GBS. CSF analysis demonstrated albuminocytological dissociation. Antibodies against GQ1b, GD1a, GD1b, GM1, GM2 and GM3 were negative. An MRI of the brain was normal. He was started on intravenous IVIg at a dosage of 2 g/kg over five days. However, there was no significant improvement, so the IVIg course was repeated after two weeks. He remained on ventilation for 75 days and had a prolonged Intensive Care Unit (ICU) stay of 80 days. His ptosis improved after four weeks. The GBS disability score at discharge on day 97 was four. He had a good recovery by the end of one year, with minor foot drop, resulting in a GBS disability score of two.
Ptosis in AMAN variant of Guillain-Barré syndrome (GBS) in case 3.
Case 4
A four-year-old boy presented with a history of voice alteration for two days and difficulty swallowing for one day. There was a history of fever with features of a respiratory infection four days prior, as well as a history of vaccination {Diphtheria-Pertussis-Tetanus (DPT) Measles-Mumps-Rubella (MMR)} two weeks earlier. On the third day of illness, he developed weakness in both lower limbs, which progressed to involve both upper limbs on the same day, requiring mechanical ventilation.
On examination, he exhibited normal higher mental functions, bilateral ptosis with no ophthalmoplegia [Table/Fig-5], bilateral lower motor neuron facial palsy and bulbar palsy. Muscle power was 3/5 in all limbs, accompanied by hypotonia and areflexia. He also showed signs of autonomic involvement, including tachycardia and excessive sweating. Complete blood count, C-reactive protein, serum electrolytes and renal and liver function tests were all normal.
Ptosis in AMAN variant of Guillain-Barré syndrome (GBS) in case 4.
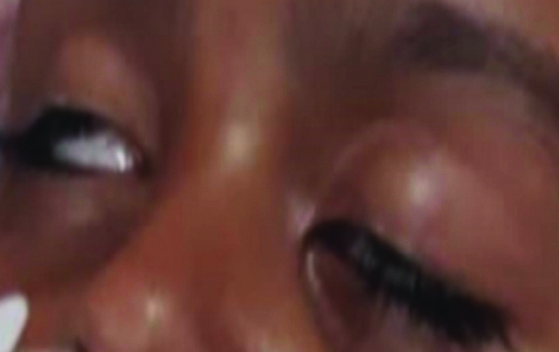
Nerve conduction studies revealed the absence of CMAP with preserved SNAP, suggestive of the AMAN variant of GBS. MRI of the brain and spine was normal. He was treated with intravenous IVIG at a dosage of 2 g/kg over five days. He was ventilated for six days and remained in the ICU for 10 days. He achieved complete recovery of ptosis by the third week. The GBS disability score at discharge on day 15 was 3. He had a complete recovery by the end of one year, with a GBS disability score of 0.
Case 5
A five-year-old girl with a history suggestive of an upper respiratory infection one week prior presented with drooping of both eyelids, difficulty swallowing and drooling of saliva, followed by weakness in both lower limbs. On examination, higher mental functions were normal, with bilateral ptosis (right more than left) [Table/Fig-6], bilateral lower motor neuron facial palsy and bulbar palsy. Power was 0/5 in all limbs, with hypotonia and areflexia. Complete blood count, C-reactive protein, serum electrolytes and renal and liver function tests were normal. Nerve conduction studies revealed an absence of CMAP with preserved SNAP, suggestive of the AMAN variant of GBS. CSF analysis showed albumino-cytological dissociation. Antibodies against GQ1b, GD1a, GD1b, GM1, GM2 and GM3 were negative. MRI of the brain and spine was normal. She was treated with intravenous IVIg at a dosage of 2g/kg over five days. She developed respiratory failure on day 5 of illness and was ventilated for 15 days. She also exhibited features of autonomic involvement, including tachycardia, hypertension and excessive sweating. Her ptosis began to improve at the beginning of the third week. The GBS disability score at discharge on day 35 was 4. She had a good recovery at the end of one year, with a GBS disability score of 1.
Ptosis in AMAN variant of Guillain-Barré syndrome (GBS) in case 5.

Discussion
The GBS is an important cause of acute flaccid weakness in children. AMAN is characterised by antibodies against the ganglioside antigens in the axolemma and infiltration of macrophages in the nodes of Ranvier. The ventral nerve root in the spinal cord is preferentially involved, resulting in axonal degeneration and conduction block [6]. The present case series illustrates an unusual clustering of AMAN variant of GBS cases with early and prominent ptosis without ophthalmoplegia and severe autonomic dysfunction. This clustering could be due to a common infectious trigger, even though we could not find evidence of any specific infection.
Ocular involvement is described in 10% of GBS cases, but ptosis without ophthalmoplegia is rarely reported [3]. Bilateral ptosis without ophthalmoplegia was reported by Talebian A et al., in a 10-year-old girl from Iran with GBS [4]. However, the nerve conduction study was not specified as AMAN in the present report. Ptosis with ophthalmoparesis was reported in a 30-year-old man by Budumuru U et al., from India with the AMAN variant of GBS and ptosis [7]. There are few case reports of ptosis without ophthalmoplegia in demyelinating GBS from adults [8,9]. To the best of authors knowledge, there are no reports of ptosis without ophthalmoplegia in the AMAN variant of GBS in children. All five cases in the present series had prominent and early ptosis but did not exhibit features of ophthalmoplegia.
At the time of presentation, the diagnostic possibilities considered were GBS, including MFS and MG. MFS is characterised by the triad of external ophthalmoplegia, ataxia and areflexia. The present cases had no features of external ophthalmoplegia or ataxia. The presence of autonomic dysfunction and features of AMAN in the nerve conduction study points against the diagnosis of MG.
Autonomic dysfunction is reported to be a common complication in the AIDP variant of GBS but is less frequently reported in AMAN [5,10]. Four of the present cases had severe autonomic dysfunction with tachycardia and hypertension and one child had frequent ventricular ectopics and was managed symptomatically. The AMAN variant of GBS is characterised by rapid progression and a severe course [11]. The study conducted by Sen S et al., from North India involving 108 GBS children observed a severe and prolonged course in the axonal variant of GBS [10]. The children had rapid progression with a severe and prolonged course and by the end of one year, all of them had good recovery.
Conclusion(s)
The AMAN variant of GBS can present with multiple cranial nerve palsies and severe autonomic dysfunction. Ptosis without ophthalmoplegia may be an early and prominent feature in children. AMAN can present with life-threatening dysautonomia. Early recognition of autonomic dysfunction is important for management.
*CMAP: Compound muscle action potential;
†SNAP: Sensory nerve action potential
[1]. Molina-Giraldo P, Ulate-Campos A, Petanàs-Argemï J, Rebollo Polo M, González-Álvarez V, Differential diagnosis of flaccid paralysis in paediatric medicineNeurol Engl Ed 2016 31(7):500-01.10.1016/j.nrleng.2014.12.009 [Google Scholar] [CrossRef]
[2]. Langille MM, Guillain-Barre Syndrome in children and adolescentsAdv Pediatr 2023 70(1):91-103.10.1016/j.yapd.2023.04.00137422300 [Google Scholar] [CrossRef] [PubMed]
[3]. Ralapanawa U, Kumarihamy P, Jayalath T, Udupihille J, Guillain-Barré syndrome with associated unilateral ptosis without ophthalmoplegia – a rare presentation: A case report and review of the literatureJ Med Case Reports 2019 13:22110.1186/s13256-019-2157-x31324211 [Google Scholar] [CrossRef] [PubMed]
[4]. Talebian A, Soltani B, Talebian M, Bilateral ptosis as the first presentation of Guillain-Barre SyndromeIran J Child Neurol 2016 10(1):70-72. [Google Scholar]
[5]. Zaeem Z, Siddiqi ZA, Zochodne DW, Autonomic involvement in Guillain–Barré syndrome: An updateClin Auton Res 2019 29(3):289-99.10.1007/s10286-018-0542-y30019292 [Google Scholar] [CrossRef] [PubMed]
[6]. Shastri A, Al Aiyan A, Kishore U, Farrugia ME, Immune-Mediated Neuropathies: Pathophysiology and ManagementInt J Mol Sci 2023 24(8):728810.3390/ijms2408728837108447 [Google Scholar] [CrossRef] [PubMed]
[7]. Budumuru U, Muralidharan K, Sowmini PR, Velayutham SS, Jeyaraj KM, Saravanan RV, AMAN with ophthalmoparesis: A rare presentationJ Assoc Physicians India 2023 71(11):103-04. [Google Scholar]
[8]. Imam YZ, Deleu D, Isolated bilateral ptosis as an early sign of Guillain-Barré SyndromeCase Rep Neurol Med 2013 2013:17829110.1155/2013/17829123585975 [Google Scholar] [CrossRef] [PubMed]
[9]. Teng HW, Sung JY, Ptosis as the initial presentation of Guillain-Barré SyndromeJ Emerg Med 2012 43(5):e283-85.10.1016/j.jemermed.2010.05.01620634019 [Google Scholar] [CrossRef] [PubMed]
[10]. Sen S, Kumar A, Roy B, Clinical Outcome of Guillain-Barré Syndrome in 108 childrenIndian Pediatr 2021 58(9):83310.1007/s13312-021-2302-733506807 [Google Scholar] [CrossRef] [PubMed]
[11]. Dimachkie MM, Barohn RJ, Guillain-Barré Syndrome and variantsNeurol Clin 2013 31(2):491-510.10.1016/j.ncl.2013.01.00523642721 [Google Scholar] [CrossRef] [PubMed]