Rapp-Hodgkin Syndrome (RHS) is a rare heterogeneous disorder characterised by ectodermal abnormalities primarily affecting the skin, teeth, eyes, hair and appendages. It is a type of Anhidrotic Ectodermal Dysplasia (AED) associated with clefts of the lip, palate and alveolus. Affected areas include the skin, hair, teeth, mouth, jaws, nails, eyes and ears. The authors present a case of RHS in a 13-year-old male who complained of deformed teeth, reduced sweating and heat intolerance. Clinical findings included sparse, thin hair on the scalp and eyebrows, sparse eyelashes, epiphora, discoloured scaly skin, a deformed pinna, microstomia, trismus, a cleft lip scar, hyponychia and a deficient maxilla. Intraoral examination showed oligodontia and misshapen teeth, along with a depapillated tongue. The patient had previously undergone cleft lip and palate surgery, ankyloblepharon release, adenoid surgery, and ear surgery at a younger age. Radiographically, there was evidence of cleft alveolus, retained deciduous teeth, impacted teeth and oligodontia. Although the patient had a normal karyotype, histopathological examination of the skin showed hyperkeratosis, acanthosis, follicular plugging, and increased pigmentation of the basal cell layer. Considering this amalgamation of signs and symptoms, a diagnosis of RHS was made, which necessitates a multidisciplinary approach for treatment. This patient will require a multidisciplinary approach to address his complaints. The authors reported the present case because it represents a rare type of ectodermal dysplasia, which is important for any physician’s knowledge due to its varied presentations.
Case Report
A 13-year-old male presented in the Outpatient Department (OPD) with spacing between his teeth, difficulty in mastication, and complaints of dark discolouration of skin, decreased sweating and intolerance to heat. Additionally, his parents noted that he exhibited facial deformities, premature greying of hair, and loss of teeth. His medical history revealed that he was a known case of RHS. Histopathological examination of a skin biopsy conducted in 2013 revealed hyperkeratosis, acanthosis, follicular plugging, hyperpigmentation and a decrease in pilosebaceous follicles, leading to the diagnosis of RHS [Table/Fig-1]. Karyotype analysis was normal [Table/Fig-2]. There was no history of similar complaints in the family. Prior to the examination, the patient provided informed consent. The patient had undergone multiple surgeries, including cleft lip surgery at seven months of age, cleft palate surgery at three years of age, ankyloblepharon release at one month of age and adenoid surgery at ten years of age.
Histopathological report showing compatibility with ectodermal dysplasia.

Karyotype analysis revealed a normal karyotype.

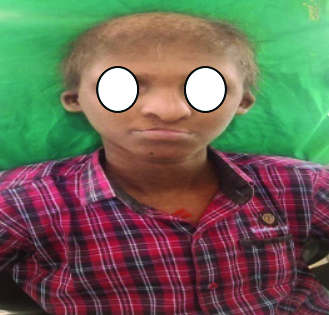
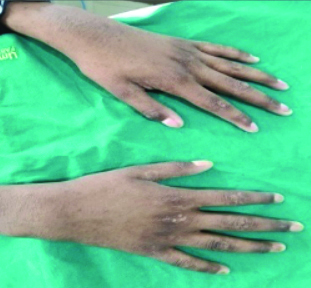
Extraoral examination revealed alopecia, scalp dermatitis, scanty grey hair, and sparse eyebrows, microtia, orofacial clefting, persistent epiphora, a depressed nasal bridge, frontal bossing, an elongated head, midfacial hypoplasia, cracked lips, dark-coloured skin and dystrophic nails [Table/Fig-3,4].
Facial profile of the patient showing light coloured hair, sparse eyelashes and eyebrows, depressed nasal bridge and thin lip.
Extremities showing dark-coloured skin and dystrophic nails.
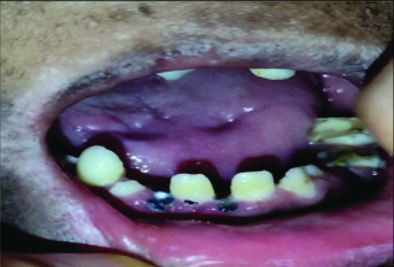
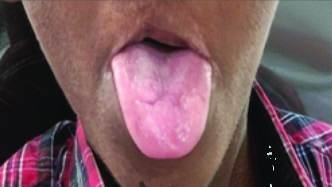
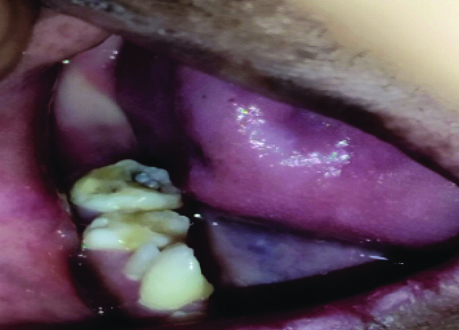
Oral examination revealed oligodontia, hypoplastic teeth and screwdriver-shaped incisors [Table/Fig-5]. There were malposed teeth with a crossbite of the maxillary teeth, delayed eruption, microglossia, a depapillated, grooved tongue, restricted mouth opening and an open mouth posture [Table/Fig-6]. A few deciduous teeth were retained and multiple decayed teeth and root stumps were noted [Table/Fig-7]. The temporomandibular joint showed no abnormalities.
Intraoral view depicting tiny conical-shaped incisors, decayed teeth and multiple root stumps.
Intraoral view revealed a depapillated tongue.
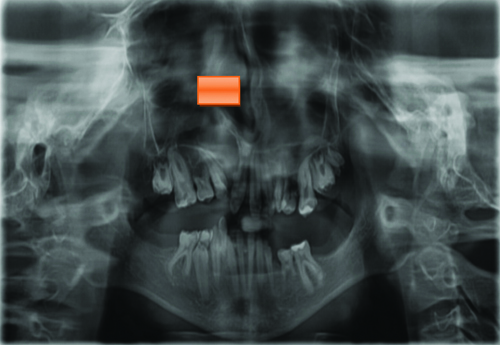
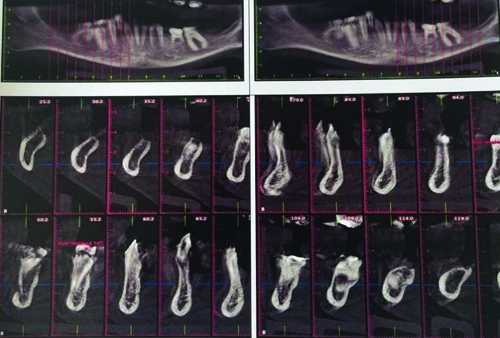
Radiographic analysis included panoramic radiography, lateral cephalogram, anteroposterior cephalogram and three-dimensional cone beam Computed Tomography (CT). The Orthopantomogram (OPG) examination revealed hypodontia, taurodontism in the mandibular right and left molar regions, a reduced enamel layer on all teeth, a retained deciduous tooth, partial anodontia, an erupting lower right premolar and a cleft alveolus [Table/Fig-8].
Intraoral view showing retained deciduous teeth.
OPG revealed a unilateral cleft alveolus (red marking), over-retained deciduous teeth and hypodontia.
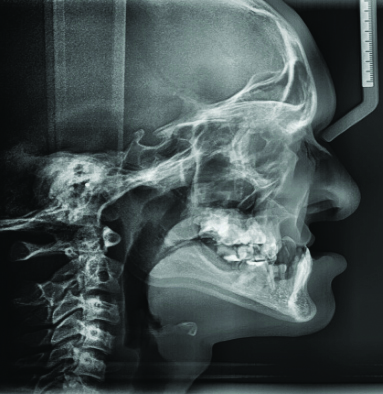
The lateral cephalogram and anteroposterior cephalogram showed midfacial hypoplasia, with a Class III tendency due to cleft-related maxillary deficiency and relative mandibular prognathism [Table/Fig-9,10]. Three-dimensional imaging of the maxilla revealed malpositioned teeth 11 and 21, labially proclined teeth 11 and 21, partial anodontia and a unilateral cleft palate with a cleft alveolus on the right side [Table/Fig-11]. In the mandibular arch, there was partial anodontia, missing tooth buds, over-retained teeth 84 and 74 and an erupting tooth 44 [Table/Fig-12].
Anteroposterior view showing showed mandibular prognathism with a Class III malocclusion.
Cephalogram revealed facial hypoplasia.
Coronal cross-section and 3-D reconstruction images revealed partial anodontia, unilateral cleft palate, and cleft alveolus.

Panoramic 3-D reconstruction and a cropped section showed partial anodontia and missing tooth buds.
A multidisciplinary approach was planned. Treatment commenced with oral hygiene instructions and dietary counselling, as the patient had widespread caries, necessitating oral prophylaxis. Pit and fissure sealants, composite restorations and root canal treatment were required. The patient was referred to a paediatric dentist. Extractions of root stumps and grossly decayed teeth were planned, followed by prosthodontic management after orthodontic consultation.
Discussion
The RHS or Rapp-Hodgkin Ectodermal Dysplasia (RHED) is a rare autosomal dominant disorder [1,2]. The term Ectodermal Dysplasias (EDs) refers to a broad and complex group of diseases that includes different clinical conditions [3,4]. They are caused by defective epidermal appendage development and are characterised by a primary deficiency in two or more of the following tissues: hair, teeth, sweat glands and nails [5]. This syndrome was first identified over thirty years ago when a case of a mother, daughter and son with AED, cleft palate, and cleft lip was reported [6]. Few authors consider RHS and Hay-Wells disease as different spectrums of the same syndrome and suggest that they can be used interchangeably [7,8]. The most common downstream missense mutation in p63 that has been identified to date is an amino acid substitution [7].
The condition RHS is regarded as one of the disorders classified into the categories of orofacial clefting and ectodermal dysplasia. The most distinguishing characteristics of RHS are hypoplastic maxilla, pili torti and cleft palate [9]. Several important clinical characteristics set RHS apart from others.
Traditionally, RHS can be distinguished from the other two ectodermal dysplasia syndromes that have a cleft palate based on clinical characteristics. Ectrodactyly-ectodermal dysplasia Cleft lip-palate (EEC) syndrome is known for the abnormal development of the middle fingers and toes, while AEC syndrome typically involves a collodion-like membrane at birth and strands of skin between the eyelids [10].
Lateral cephalometric analysis has shown midfacial hypoplasia to be a feature of RHS [10]. This is a very important finding, as the assessment of current and proposed orofacial development affects the dental treatment plan.
Individuals with RHS may show symptoms such as reduced sweat glands, sparse hair on the scalp and eyebrows, thin eyelashes, excessive tearing, outward-rolling eyelids, discoloured scaly skin, abnormal ear shape with cleft lip and palate and underdeveloped nails. Moreover, extraoral findings consist of an elongated skull, lips that cannot close properly, a misshapen nose, wide-set eyes, and a prominent ridge above the eye sockets. Typically, mental retardation and sensory-neural deafness are not present [6]. Intraoral examination generally reveals malocclusion, microglossia, oronasal fistula due to alveolar cleft, oligodontia, hypoplastic teeth, multiple retained teeth, caries and multiple impacted teeth. A familial tendency may be noted [10]. Radiographic examination generally corroborate with the clinical features like retruded cleft maxilla, relatively prognathic mandible, alveolar cleft, hypoplastic teeth, malformed enamel layer, over-retained deciduous teeth, impacted teeth and carious teeth [10]. This patient exhibited dental and general findings that were similar to the aforementioned signs and symptoms.
There is significant overlap between RHS and other ED, such as ankyloblepharon-ectodermal dysplasia-clefting (AEC, also called Hay-Wells syndrome) and EEC syndrome [12]. AEC syndrome is characterised by the aberrant development of median rays in the hands and feet, syndactyly, as well as, ankyloblepharon filiforme adenatum, which refers to strands of skin between the eyelids, and a collodion-like membrane at birth. Some authors consider RHS and Hay-Wells disease to be different spectrums of the same syndrome and use the terms interchangeably [13].
To treat this syndrome, a multidisciplinary team is required, including a dentist, dermatologist, maxillofacial surgeon, otolaryngologist, plastic surgeon, ophthalmologist, paediatrician, audiologist, psychiatrist, speech therapist and others. It is necessary to reassure and instruct parents about the low rate of mental retardation [14]. Treatment options include surgeries for cleft lip and palate, ear tube placement, ankyloblepharon release and maintenance of skin hygiene.
From a dental perspective, treatment may involve secondary alveolar bone grafting, repair of carious teeth, extraction of hopeless teeth, replacement of missing teeth using prosthetics and/or dental implants, coverage of hypoplastic hard tissue, and the use of salivary replacements, if dry mouth worsens [15].
Conclusion(s)
In conclusion, the psychological effects of tooth loss, hair loss and facial deformity on these patients are the most important aspects to consider. Therefore, early diagnosis and a stepwise treatment plan with proper dental therapy are important factors in managing RHS.
[1]. Plottova-Puech I, Vidal C, Godard W, Cambazard F, Rapp-Hodgkin’s Syndrome: Two casesAnn Dermatol Venereol 2003 130:331-36. [Google Scholar]
[2]. Rapp RS, Hodgkin WE, Anhidrotic ectodermal dysplasia: Autosomal dominant inheritance with palate and lip anomaliesJ Med Genet 1968 5:269-72.10.1136/jmg.5.4.2695713637 [Google Scholar] [CrossRef] [PubMed]
[3]. Pinheiro M, Freire-Maia N, Ectodermal dysplasias: A clinical classification and a causal reviewAm J Med Genet 1994 53:153-62.10.1002/ajmg.13205302077856640 [Google Scholar] [CrossRef] [PubMed]
[4]. Priolo M, Silengo M, Lerone M, Ravazzolo R, Ectodermal dysplasias: Not only ‘skin’ deepClin Genet 2000 58:415-30.10.1034/j.1399-0004.2000.580601.x11149610 [Google Scholar] [CrossRef] [PubMed]
[5]. Lamartine J, Towards a new classification of ectodermal dysplasiasClin Exp Dermatol 2003 28:351-55.10.1046/j.1365-2230.2003.01319.x12823289 [Google Scholar] [CrossRef] [PubMed]
[6]. Chatterjee M, Neema S, Mukherjee S, Rapp hodgkin syndromeIndian Dermatol Online J 2017 8(3):215-16.10.4103/idoj.IDOJ_100_1628584763 [Google Scholar] [CrossRef] [PubMed]
[7]. Kantaputra PN, Hamada T, Kumchai T, McGrath JA, Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectoder mal dysplasiaJ Dent Res 2003 82:433-37.10.1177/15440591030820060612766194 [Google Scholar] [CrossRef] [PubMed]
[8]. Clements SE, Techanukul T, Holden ST, Mellerio JE, Dorkins H, Escande F, Rapp-Hodgkin and Hay-Wells ectodermal dysplasia syndromes represent a variable spectrum of the same genetic disorderBr J Dermatol 2010 163(3):624-29.10.1111/j.1365-2133.2010.09859.x20491771 [Google Scholar] [CrossRef] [PubMed]
[9]. Kannu P, Savarirayan R, Ozoemena L, White SM, McGrath JA, Rapp-Hodgkin ectodermal dysplasia syndrome: The clinical and molecular overlap with Hay-Wells syndromeAm J Med Genet A 2006 140(8):887-91.10.1002/ajmg.a.3118716532463 [Google Scholar] [CrossRef] [PubMed]
[10]. Karthikeyani S, Thirumurthy VR, Yuvaraja BA, Dental management of Rapp-Hodgkin syndrome associated with oral cleft and hypodontiaJ India Soc Pedod Prev Dent 2016 34:192-95.10.4103/0970-4388.18045327080974 [Google Scholar] [CrossRef] [PubMed]
[11]. Sahin MT, Türel-Ermertcan A, Chan I, McGrath JA, Oztürkcan S, Ectodermal dysplasia showing clinical overlap between AEC, Rapp Hodgkin and CHAND syndromesClin Exp Dermatol 2004 29:486-88.10.1111/j.1365-2230.2004.01584.x15347331 [Google Scholar] [CrossRef] [PubMed]
[12]. Hay RJ, Wells RS, The syndrome of ankyloblepharon, ectodermal defects and cleft lip and palate: An autosomal dominant conditionBr J Dermatol 1976 94:277-89.10.1111/j.1365-2133.1976.tb04384.x946410 [Google Scholar] [CrossRef] [PubMed]
[13]. Gupta S, Mittal S, Tewari S, Nandal S, Ectrodactyly, ectodermal dysplasia, and cleft lip/palate syndrome: A case reportInternational Journal of Health Sciences and Research 2017 7(4):459-62. [Google Scholar]
[14]. Elhamouly Y, Dowidar KM, Dental management of a child with ectrodactyly ectodermal dysplasia cleft lip/palate syndrome: A case reportSpec Care Dentist 2019 39(2):236-40.10.1111/scd.1236430720215 [Google Scholar] [CrossRef] [PubMed]