Familial Adenomatous Polyposis: An Uncommon Autosomal Genetic Condition
Simran Khan1, Shreya Giri2, Suhit Naseri3
1 Junior Resident, Department of Pathology, Datta Meghe Institute of Medical Sciences, Sawangi (Meghe), Maharashtra, India.
2 Junior Resident, Department of Pathology, Datta Meghe Institute of Medical Sciences, Sawangi (Meghe), Maharashtra, India.
3 Junior Resident, Department of Pathology, Datta Meghe Institute of Medical Sciences, Sawangi (Meghe), Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Simran Khan, Junior Resident, Department of Pathology, Jawaharlal Nehru Medical College, Sawangi (Meghe)-442004, Maharashtra, India.
E-mail: sabhatsimrankhan416@gmail.com
Anal mass, APC gene, Colon cancer, Histopathology
A 40-year-old man presented to the Department of Emergency Medicine with severe pain in abdomen which was present for the past two days. He experienced three episodes of vomiting and two episodes of diarrhoea which was greenish in colour without any blood or mucus. A mass from the anal region was also noticed while defecating which was insidious in onset and gradually progressing in size since one month. Physical examination upon deep palpation revealed generalised abdominal pain but no signs of peritoneal involvement. He claimed to have lost weight during the last four months. Notably, his family history included several relatives who had intestinal polyps and colon cancer. Laboratory findings indicated 8.5 g/dL haemoglobin levels and a mean corpuscular volume of 70 fL, along with a positive faecal occult blood test. Normal serum levels of electrolytes, creatinine, glucose, haemoglobin, and pH were observed. A differential diagnosis of haemorrhoids and rectal ulcers was made.
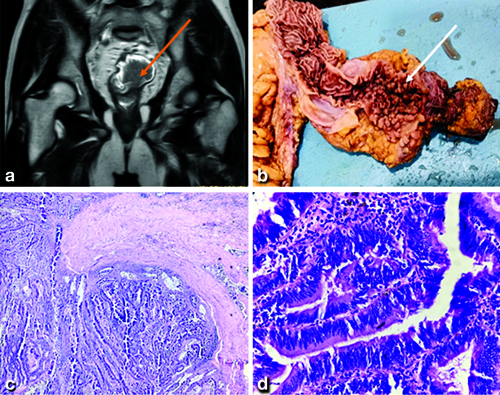
Following an MRI pelvis, coronal T2W1 showed a cauliflower-shaped polypoidal mass lesion noted in the rectum appearing heterogeneously hypointense on T2W1 [Table/Fig-1a]. A total proctolectomy was performed based on the imaging findings. Exploration was performed by midline vertical incision and resectability was assessed by noting the relationship of the mass with the sacrum, ureters, urinary bladder, and prostate. The disease spread was also assessed by examination of the liver, peritoneum, and evidence of ascites. Differential diagnosis of gastritis, attenuated Familial Adenomatous Polyposis (FAP), hamartomatous polyp, hyperplastic polyposis, hereditary mixed polyposis syndrome, and lynch syndrome was made. The specimen was then sent for histopathological diagnosis. The specimen on cut-section revealed multiple pedunculated (approximately, 165 polyps, of size less than 4 mm) polypoidal growths located on the circumferential walls of the ascending colon, transverse colon, and descending colon [Table/Fig-1b]. Microscopic findings with haematoxylin-eosin staining on X40 [Table/Fig-1c] showed conserved crypt architecture and increased number of glands with moderate dysplasia and X400 [Table/Fig-1d] demonstrated colonic epithelium showing columnar cells with reduced mucin content, hyperchromatic nuclei, and frequent mitotic figures along with moderatedysplasia, indicative of histopathological characteristics associated with FAP. The time following surgery was uneventful. The patient was discharged after 15 days and was advised to follow-up in surgical oncology OPD after one week.
a) MRI pelvis (Coronal T2W1), a cauliflower-shaped polypoidal mass in the rectum; b) multiple pedunculated polypoidal growths located on the circumferential wall of the colon; c) Haematoxylin-eosin staining on X40 shows conserved crypt architecture and an increased number of glands with moderate dysplasia; d) Haematoxylin-eosin staining on X400 demonstrates colonic epithelium showing columnar cells with reduced mucin content, hyperchromatic nuclei, and frequent mitotic figures, indicative of histopathological characteristics associated with Familial Adenomatous Polyposis (FAP).
Adenomatous Polyposis Coli (APC) gene on chromosome 5q21 is the causative agent of FAP, an uncommon autosomal dominant hereditary cancer-predisposition condition [1]. It is an autosomal dominant disease in which the large bowel develops more than 100 adenomatous polyps [2]. During metaphase, the APC gene acts as a tumour suppressor and is crucial for proper chromosomal alignment. In colonic epithelial cells, the APC protein promotes apoptosis. However, mutations in the APC protein disrupt apoptosis and allow unchecked cell growth, leading to the formation of adenomas. The progression from adenoma to cancer is well-documented, beginning with the inactivation of the APC gene. Subsequent mutations in other oncogenes and tumour suppressor genes, such as p53 and KRAS, lead to dysplasia and eventually cancer. Mutations in the APC gene are common in both sporadic and familial cases of Colorectal Cancer (CRC) [3]. If left untreated, almost all patients with the diagnosis of FAP develop colorectal carcinoma which is usually diagnosed between 32-50 years of age. FAP affects both sexes equally at birth, has an incidence of around 1/8,300, and causes fewer than 1% of instances of CRC. Prevalence in the European Union is thought to be between 1/11,300 and 37,600. Until the adenomas are big and numerous, causing rectal bleeding or even anaemia, or until cancer occurs, the majority of patients remain asymptomatic for years. Cancers often begin to manifest ten years after the polyps first arise [4]. Although, there has been much written about the relationships between genotype and phenotype, there is still diversity within and across families. Determining the causal pathogenetic mutation in every pedigree is essential for creating a cancer management programme and a follow-up plan for at-risk families [5]. In addition, patients with FAP have an increased chance of acquiring additional cancers, such as hepatoblastoma, thyroid cancer, gastric cancer, and duodenal or ampullary cancer [6,7]. Malignant extracolonic manifestations can cause the patient’s death, despite the fact that they are frequently benign. Once an afflicted person’s hereditary CRC syndrome is recognised, monitoring needs to be carried out on both that person and any at-risk family members [8]. For patients with the APC mutation, lifetime screening is advised [9]. When choosing a therapy and monitoring approach for patients with FAP, good patient compliance is crucial in determining the patients’ prognosis [10].
This case underscores the rarity and complexity of diagnosing FAP. This rare variant poses significant diagnostic challenges due to its infrequent occurrence and atypical radiological and histological features [7,9]. [Table/Fig-2] shows the published case of AFP. In conclusion, recognising the uncommon and atypical presentations of FAP is crucial for accurate diagnosis and appropriate management.
Published cases on Familial Adenomatous Polyposis (FAP) [7,9].
| Authors | Year ofpublication | Place of study | No. of cases | Findings of the case |
|---|
| Righetti AEM et al., [7] | 2011 | Brazil | 29-year-old female | FAP diagnosis |
| Adla A et al., [9] | 2023 | USA | 21-year-old male | FAP diagnosis |
| Present case | 2024 | India | 40-year-old male | FAP diagnosis |
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Jul 06, 2024
Manual Googling: Aug 07, 2024
iThenticate Software: Aug 08, 2024 (9%)
[1]. Beech D, Pontius A, Muni N, Long W, Familial adenomatous polyposis: A case report and review of the literatureJ Natl Med Assoc 2001 93:208-13. [Google Scholar]
[2]. Campos FG, Imperiale AR, Seid VE, Perez RO, Da Silva E Sousa AH, Kiss DR, Rectal and pouch recurrences after surgical treatment for familial adenomatous polyposisJ Gastrointest Surg 2009 13(1):129-36.10.1007/s11605-008-0606-818766422 [Google Scholar] [CrossRef] [PubMed]
[3]. Menon G, Carr S, Kasi A, Familial adenomatous polyposisStatPearls 2022 StatPearls PublishingAvaialble from: http://www.ncbi.nlm.nih.gov/books/NBK538233 [Google Scholar]
[4]. Half E, Bercovich D, Rozen P, Familial adenomatous polyposisOrphanet Journal of Rare Diseases 2009 4:01-23.10.1186/1750-1172-4-2219822006 [Google Scholar] [CrossRef] [PubMed]
[5]. Cerasuolo A, Miele E, Russo M, Aversano A, Cammarota F, Duraturo F, Sporadic pediatric severe familial adenomatous polyposis: A case reportMol Clin Oncol 2020 13(3):0110.3892/mco.2020.209032754334 [Google Scholar] [CrossRef] [PubMed]
[6]. Chaves JJ, Chaves-Cabezas V, Parra-Medina R, Chaves-Chamorro JO, Familial adenomatous polyposis: case report and literature reviewCureus 2022 14(11):e3160910.7759/cureus.31609 [Google Scholar] [CrossRef]
[7]. Righetti AEM, Jacomini C, Parra RS, De Almeida ALNR, Rocha JJR, Féres O, Familial adenomatous polyposis and desmoid tumorsClinics 2011 66(10):1839-42.10.1590/S1807-5932201100100002722012061 [Google Scholar] [CrossRef] [PubMed]
[8]. Anaya DA, Chang GJ, Rodriguez-Bigas MA, Extracolonic manifestations of hereditary colorectal cancer syndromesClin Colon Rectal Surg 2008 21(04):263-72.10.1055/s-0028-108994120011437 [Google Scholar] [CrossRef] [PubMed]
[9]. Adla A, Zheng M, Singhal M, An unsuspecting case of Familial Adenomatous Polyposis (FAP)Cureus 2023 15(4):e3835210.7759/cureus.3835237266046 [Google Scholar] [CrossRef] [PubMed]
[10]. Dalavi SB, Vedpalsingh TH, Bankar SS, Ahmed MH, Bhosale DN, Familial Adenomatous Polyposis (FAP)- A case study and review of literatureJ Clin Diagn Res 2015 9(3):PD0510.7860/JCDR/2015/11636.569625954663 [Google Scholar] [CrossRef] [PubMed]