Among fungal infections, IPA is one of the leading infectious causes of death in immunosuppressed and immunocompromised patients, particularly following bone marrow transplants, stem cell transplants, and in leukaemia patients, with a mortality rate of around 38.5% [9-11]. Among Aspergillus species, Aspergillus fumigatus has been identified as the predominant agent of IPA, followed by A. flavus, A. terreus, and A. niger. Routine mycological identification is typically based on microscopic and cultural characteristics; however, the recent increase in cryptic or rare species, along with the presence of multidrug resistance in some of these species, underscores the need for identification at the species level [12-15].
Since the species-level identification of morphologically indistinguishable cryptic or sibling species has become difficult, molecular methods such as MLSA have been employed [16]. The frequency of these sibling species was previously reported as 11% in the Transplant-Associated Infection Surveillance Network (TRANSNET) study and 15% in the FILPOP (Population-grounded survey of filamentous fungi) study [17,18]. Various molecular methods using Polymerase Chain Reaction (PCR) have been developed for species-level identification, as well as for identifying previously unknown and rare species. In certain scenarios, the direct identification of Aspergillus from clinical samples, such as BAL and blood samples, has also been made possible using real-time PCR [19,20]. Some cryptic species may also exhibit reduced susceptibility to multiple antifungal agents, such as A. calidoustus and A. lentulus. Antifungal susceptibility testing using Clinical and Laboratory Standards Institute (CLSI) and European Committee on Antimicrobial Susceptibility Testing (EUCAST) will be useful in determining MIC values, which further aids in identifying drug-resistant species [20-22]. Thus, accurate species-level identification is crucial for establishing successful treatment [19].
The primary objective of the study was to conduct species-level identification of Aspergillus isolates from clinical samples using MLSA and to conclude the ubiquity and distribution of both common and cryptic Aspergillus species in the clinical setting of Chennai, Southern India. The secondary objective was to compare the efficacy of traditional morphological methods with molecular techniques in identifying Aspergillus species, as well as to evaluate the clinical significance of cryptic Aspergillus species, particularly in terms of their antifungal susceptibility profiles and implications for patient management and treatment outcomes.
Materials and Methods
This descriptive cross-sectional study was conducted over a period of eight months in three tertiary care centres in Chennai, Southern India. The study commenced in January 2023 and concluded in August 2023. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board (REF:IEC-NI/18/SEP/66/64 & IEC-CS App. No.: AMH-006/10-18). All patients provided informed consent for the utilisation of their clinical samples in this investigation.
Inclusion criteria: Patients with clinical symptoms suggestive of aspergillosis, patients with positive culture growth of Aspergillus species from clinical samples were included in the study.
Exclusion criteria: Patients who failed to provide informed consent, samples contaminated with other microorganisms were excluded from the study.
Sample size: The sample size of 60 Aspergillus strains was determined based on the availability of clinical samples during the study period. This study was time-bound, and all available subjects meeting the inclusion criteria were considered.
Sixty Aspergillus strains were subsequently isolated from clinical samples collected at three tertiary care centres: Sri Ramachandra Institute of Higher Education and Research, Apollo Hospitals, Gream’s Road, and Apollo Speciality Hospital, Teynampet, in Chennai, Tamil Nadu, India. The patients with Aspergillus growth were classified as having proven, possible, or probable aspergillosis based on the EORTC/MSG criteria [23].
Study Procedure
Phenotypic identification: The Aspergillus isolates were cultured on SDA and oatmeal agar (both from HI Media Laboratories Pvt. Ltd., Mumbai, India), supplemented with antibiotics. Cultures were incubated at two different temperatures, 37°C and 25°C. Routine examinations of the culture tubes were conducted regularly to monitor the presence of growth. Initial phenotypic identification of Aspergillus species was performed based on physiological characteristics such as temperature requirements and growth rate, as well as various colony morphological features. Microscopic identification was carried out using either the direct culture method or the slide culture technique, with Lactophenol Cotton Blue (LPCB) mounts.
Molecular identification: The isolated Aspergillus strains underwent molecular identification. Genomic fungal DNA was extracted using an in-house phenol-chloroform method standardised in the laboratory. The extracted DNA was amplified using specific primers for the ITS, beta-tubulin, hydrophobin, and calmodulin genes. Amplified gene fragments were sequenced utilising the Big Dye Terminator V.3.1 cycle sequencing kit at Eurofins Genomics India Pvt. Ltd., Bangalore, Karnataka, India.
Multilocus Sequence Analysis (MLSA): MLSA is a powerful molecular technique used for the precise identification and phylogenetic analysis of microorganisms. It involves sequencing multiple housekeeping genes, which are essential and conserved genes found in all cells, to compare genetic similarities and differences among species. MLSA provides high-resolution discrimination between closely related species, offering a more accurate and reliable method for identifying and classifying microorganisms compared to single-gene analyses. This technique is particularly useful in studying cryptic species and in cases where phenotypic methods are insufficient for accurate identification [20].
DNA extraction: DNA extraction from Aspergillus was performed using an in-house phenol-chloroform method that has been standardised in our laboratory. A loopful of fungal growth was scraped from a two-day-old culture grown on oatmeal/SDA and finely ground with a sterile mortar and pestle containing 0.5 mL of lysis buffer.
Preparation of lysis buffer: The lysis buffer was prepared with 10 mM Tris base (pH 8), 1 mM Ethylenediaminetetraacetic Acid (EDTA), 100 mM sodium chloride, and 3% lauryl sulphate in 100 mL of distilled water. After grinding, the mould was transferred to a 1.5 mL Eppendorf tube and incubated at 56°C for half an hour after adding 10 μL of proteinase K. The suspension was then vortexed and incubated at 100°C in a water bath for one minute. Next, 500 μL of a 1:1 mixture of phenol and chloroform was added, followed by vortexing. The mixture was centrifuged at 10,000 Rotations Per Minute (rpm) for 10 minutes. In another microcentrifuge tube, 500 μL of chloroform was added, vortexed, and then centrifuged at 10,000 rpm for 10 minutes. Subsequently, 500 μL of cold isopropyl alcohol was added, followed by centrifugation at 10,000 rpm for 10 minutes to precipitate the DNA. The supernatant was discarded, and 70% ethanol was added to the precipitated DNA. The mixture was vortexed and centrifuged at 10,000 rpm for 10 minutes. After discarding the supernatant, the pellet was allowed to air dry for 30 minutes. Finally, the pellet was eluted by suspending it in 50 μL of sterile nuclease-free water or TE buffer. The eluted DNA can be stored at -20°C for further use. The positive control strains used were Aspergillus flavus ATCC 16883 and Aspergillus fumigatus ATCC 1022.
PCR amplification: The primer pairs for specific genes used in DNA amplification and sequencing included Internal Transcribed Spacer (ITS1 and ITS4), Hydrophobin (rod A1 and rod A2), Calmodulin (cmd5 and cmd6), and Beta-tubulin (Bt2a and Bt2b) genes. The primer pairs for the partial regions of four genes used for DNA amplification are summarised in [Table/Fig-1]. Negative (non-template) and positive controls were included in each amplification reaction. The PCR conditions for each gene are detailed in [Table/Fig-2,3,4 and 5]. This study utilised PCR amplification to identify Aspergillus species. Specifically, conventional PCR was employed, targeting the selected genes: ITS, beta-tubulin, hydrophobin, and calmodulin. The PCR reactions were standardised for each gene, and the amplified gene fragments were sequenced to confirm species-level identification.
Showing genes and their primer pairs used for DNA amplification.
| Gene sequences | Primer pairs | Primer sequence (5’-3’) |
|---|
| Internal transcribed spacer | ITS1 F | TCCGTAGGTGAACCTGCGG |
| ITS4 R | TCCTCCGCTTATTGATATG |
| Beta-tubulin | Bt2a F | AATAGGTGCCGCTTTCTGG |
| Bt2b R | AGTTGTCGGGACGGAAGAG |
| Hydrophobin | rodA1 F | GCTGGCAATGGTGTTGGCAA |
| rodA2 R | AGGGCAATGCAAGGAAGACC |
| Calmodulin | cmd5 F | CCGAGTACAAGGAGGCCTTC |
| cmd6 R | CCGATAGAGGTCATAACGTGG |
Forward primer demarcated as F; Reverse primer demarcated as R
Showing PCR reactions for Internal Transcribed Spacer (ITS) gene.
| Reaction | Temperature | Time |
|---|
| Initial denaturation | 95°C | 5 min |
| Denaturation | 95°C | 1 min |
| Annealing | 56°C | 1 min |
| Extension | 72°C | 2 min |
| Final extension | 72°C | 5 min |
| Number of cycles-35 | |
Showing PCR reaction for Beta-tubulin gene.
| Reaction | Temperature | Time |
|---|
| Initial denaturation | 94°C | 5 min |
| Denaturation | 94°C | 1 min |
| Annealing | 64°C | 1 min |
| Extension | 68°C | 2 min |
| Final extension | 68°C | 5 min |
| Number of cycles-35 | |
Showing PCR reaction for calmodulin gene.
| Reaction | Temperature | Time |
|---|
| Initial denaturation | 95°C | 5 min |
| Denaturation | 95°C | 1 min |
| Annealing | 54°C | 30 sec |
| Extension | 72°C | 1 min |
| Final extension | 72°C | 5 min |
| Number of cycles-35 | |
Showing PCR reaction for hydrophobin gene.
| Reaction | Temperature | Time |
|---|
| Initial denaturation | 95°C | 5 min |
| Denaturation | 95°C | 30 sec |
| Annealing | 53°C | 30 sec |
| Extension | 72°C | 1 min |
| Final extension | 72°C | 5 min |
| Number of cycles-35 | |
Phylogenetic analysis: Sequences were verified manually using BioEdit and MEGA X software and aligned using CLUSTAL W software. The Molecular Evolutionary Genetics Analysis software (MEGA X) was utilised for the phylogenetic analysis of the ITS, beta-tubulin, hydrophobin, and calmodulin gene sequences. The analysis of multiple gene sequences was performed using various methods, including maximum likelihood, maximum parsimony, and neighbour-joining, with 1000 bootstrap replications. The evolutionary relationships among the various Aspergillus species were analysed using these sequences. Closely related species were matched with known reference strain sequences obtained from the NCBI GenBank database. The genes and their corresponding primer pairs used for DNA amplification are provided in [Table/Fig-1] [20].
PCR reactions (50 μL) were performed in a thermocycler. The standardisation of PCR reactions for all four genes is shown in [Table/Fig-2,3,4 and 5], respectively. After gel purification, the amplified gene fragments of the isolated Aspergillus species were sequenced using the Big Dye Terminator V.3.1 cycle sequencing kit at Eurofins Genomics India Pvt. Ltd., Bangalore, Karnataka, India [24]. The obtained nucleic acid sequences were subjected to the Basic Local Alignment Search Tool (BLAST) algorithm for nucleotide searches and comparative analysis at the National Centre for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/BLAST/). For the final analysis, sequences that matched more than 99% were considered and then compared with selected culture collection reference strains.
Sequences were manually verified using BioEdit and MEGA X software and aligned with CLUSTAL W software. The evolutionary relationships of various Aspergillus species were analysed using MEGA X software through methods such as maximum likelihood, maximum parsimony, and neighbour-joining, with 1000 bootstrap replications. The sequences were compared to reference strains from the NCBI GenBank database.
Statistical Analysis
The statistical analysis was conducted using The statistical analysis was conducted using Statistical Package for the Social Sciences (SPSS) software version 25.0 (IBM, Armonk, NY, USA). Descriptive statistics were employed to summarise the data. Bootstrap analysis was used in the phylogenetic analysis to ensure the robustness of the tree [25].
Results
Out of 60 isolated Aspergillus specimens, 26 (43.3%) belonged to the Aspergillus flavus complex, 16 (26.7%) to the Aspergillus fumigatus complex, 12 (20%) to the Aspergillus niger complex, and 6 (10%) to the Aspergillus terreus complex. The initial speciation of Aspergillus was based on microscopic morphology and cultural characteristics. These Aspergillus isolates were obtained from various clinical specimens, including bronchial wash 6 (10%), BAL fluid 10 (16.7%), sputum 3 (5%), ear swab 20 (33.33%), endotracheal aspirate 8 (13.3%), pus and tissue from the nasal cavity and paranasal sinus 11 (18.3%), and tissue from other sites 2 (3.3%), as shown in [Table/Fig-6].
Showing the number of various clinical specimens from which Aspergillus species were isolated.
| S. No. | Specimen | n (%) |
|---|
| 1 | Bronchial wash | 6 (10) |
| 2 | Bronchoalveolar Lavage (BAL) | 10 (16.7) |
| 3 | Sputum | 3 (5) |
| 4 | Ear swab | 20 (33.3) |
| 5 | Endotracheal aspirate | 8 (13.3) |
| 6 | Nasal cavity and paranasal sinus | 11 (18.3) |
| 7 | Others | 2 (3.3) |
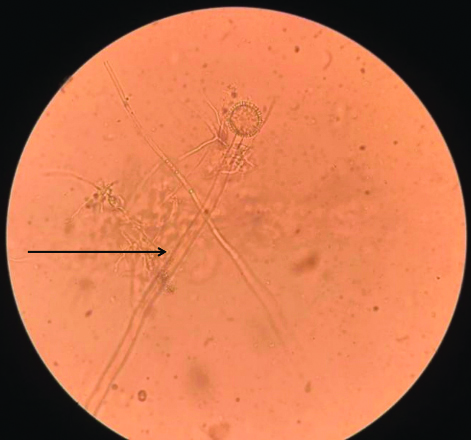
[Table/Fig-7] shows narrow septate hyphae stained with 10% KOH and visualised under a light microscope at 400x magnification, highlighting the characteristic fungal structures present in the sample.
KOH from the sample showing narrow septate hyphae at 400x magnification.
[Table/Fig-8] displays the subculture and maintenance of Aspergillus species on SDA and oatmeal agar. The image includes distinct colonies of Aspergillus, each exhibiting their characteristic growth patterns and morphology.
Subculture and maintenance of stock culture on SDA/Oat meal Agar. a) Aspergillus terreus; b) Aspergillus flavus; c) Aspergillus niger; d) Aspergillus fumigatus.

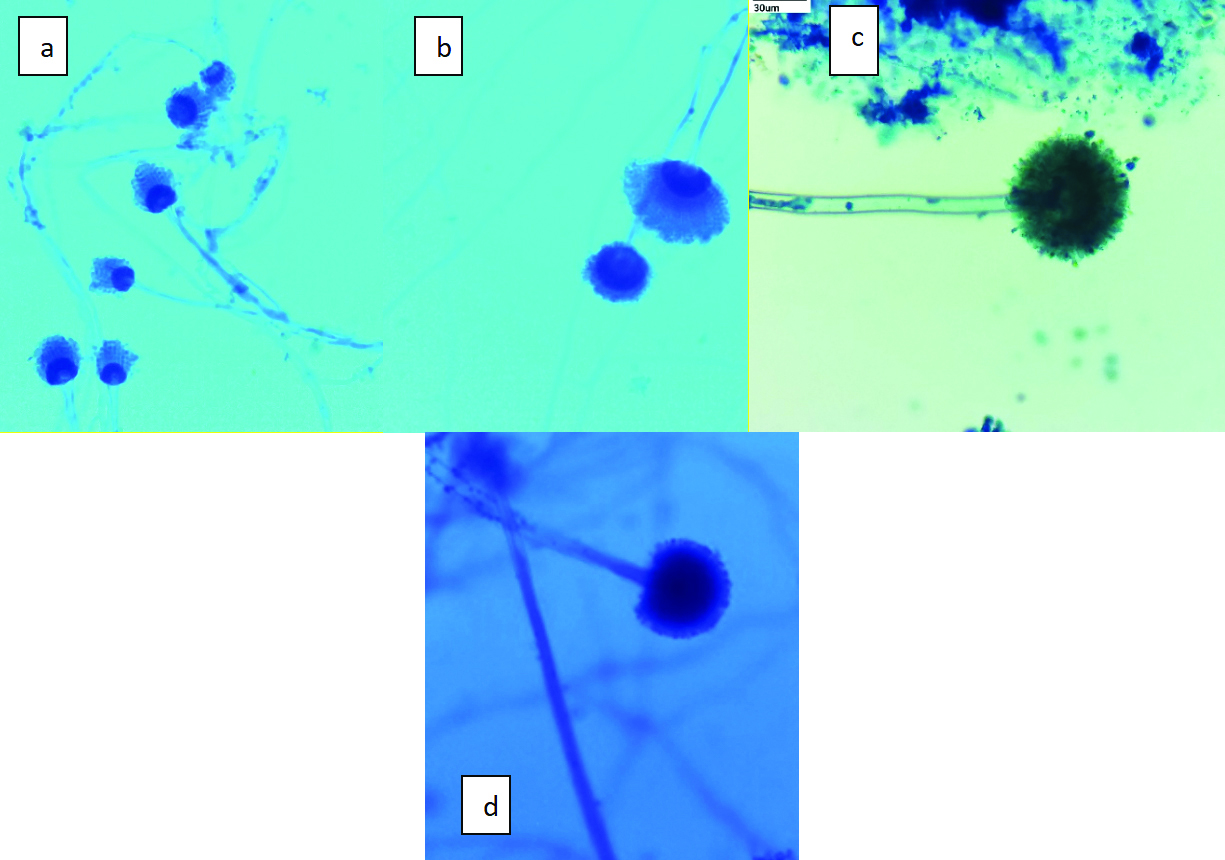
[Table/Fig-9] shows phenotypic identification of Aspergillus species using an LPCB mount. The colonies in [Table/Fig-8] are presented in macroscopic view on the agar plates, with no additional staining applied, and are photographed at standard plate observation magnification, typically around 1x-2x. In contrast, [Table/Fig-9] presents the phenotypic identification of various Aspergillus species using LPCB staining. This figure highlights the distinctive microscopic features of species such as Aspergillus fumigatus, Aspergillus terreus, Aspergillus niger, and Aspergillus flavus. The samples are stained with LPCB and visualised under a light microscope at 400x magnification, which enhances the visibility of hyphal structures and conidiophores. These updates to the figures were made to ensure that the visual data is as accurate and informative as possible, thereby enhancing the overall quality and reliability of the study.
In [Table/Fig-10], the DNA fragments analysed in this experiment are from the beta-tubulin gene of Aspergillus, with an expected size range of 600 to 700 base pairs (bp).
The images show the microscopic features in LPCB mount a) Aspergillus fumigatus; b) Aspergillus terreus; c) Aspergillus niger; d) Aspergillus flavus (All 400x).
Agrose gel electrophoresis.
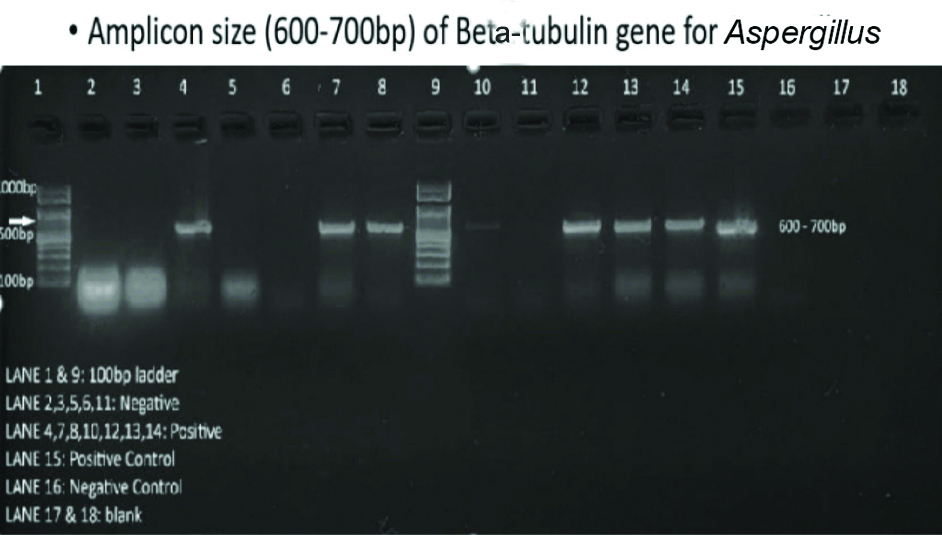
The presence of bands in the expected size range indicates the presence of the target DNA sequence in the sample. This gel electrophoresis technique is widely used in molecular biology for various applications, including DNA fingerprinting, genetic disease diagnosis, and the identification of genetic material in both research and clinical settings.
The MLSA was conducted to identify Aspergillus species at the molecular level. This analysis revealed a diverse range of Aspergillus species, including common ones like A. flavus, A. fumigatus, A. terreus, and A. niger. Additionally, rare species such as A. awamori, A. carneus, A. lentulus, A. oryzae, A. tamarii, A. tubingensis, and A. welwitschiae were identified. The MLSA process involved DNA extraction, PCR amplification of selected genes (ITS, beta-tubulin, hydrophobin, and calmodulin), sequencing, and phylogenetic analysis. The results were compared with reference strains, providing valuable insights into the genetic diversity of Aspergillus species in clinical samples.
The MLSA results revealed a high sequence identity of 99% to 100% with various Aspergillus species during BLAST analysis. Rare species such as A. awamori (n=2, 3.3%), A. carneus (n=1, 1.7%), A. lentulus (n=1, 1.7%), A. oryzae (n=3, 5%), A. tamarii (n=2, 3.3%), Aspergillus tubingensis (n=2, 3.3%), and A. welwitschiae (n=2, 3.3%) were also identified. These rare isolates exhibited similar micro- and macro-morphology to their corresponding Aspergillus species complexes. Most isolates displayed the typical and characteristic morphology of specific Aspergillus species when grown at both 25°C and 37°C. However, some rare isolates appeared as non sporulating, white colonies with atypical morphological features. These isolates were initially misidentified during phenotypic classification.
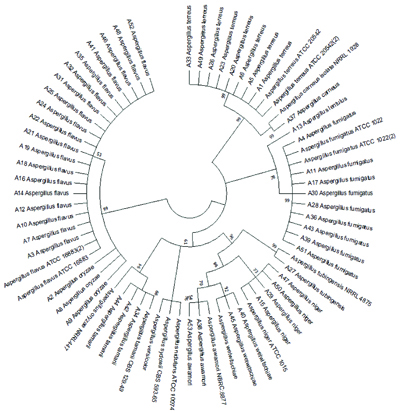
Phylogenetic analysis using ITS, beta-tubulin, hydrophobin, and calmodulin genes was conducted employing maximum parsimony, maximum likelihood, and neighbour-joining methods with 1000 bootstrap replications. The results were depicted in a dendrogram, as shown in [Table/Fig-11], illustrating the phylogenetic relationships among the isolated Aspergillus species.
Showing phylogenetic relationship between isolated Aspergillus species using Maximum likelihood method- 1000 bootstrap replicas.
Discussion
The present investigation delivers valuable insights into the diversity and prevalence of Aspergillus species in clinical samples from Chennai, Southern India, using MLSA. Present study findings showed a diversity of Aspergillus species, with A. flavus and A. fumigatus, along with A. terreus and A. niger, being the most common species. These results are consistent with previous studies conducted in different geographical areas [11]. The use of MLSA allowed for more accurate species identification, revealing not only common but also rare species such as A. awamori, A. carneus, and A. tubingensis. This highlights the importance of using molecular techniques to accurately characterise fungal isolates, especially those with non specific physiological features. In comparison, present study showed a higher proportion of Aspergillus terreus compared to some previous studies [12], indicating a possible regional variation in the ubiquity of Aspergillus species. In contrast, reports from various other countries, including the United States of America, Brazil, the Middle East, and Europe, have predominantly identified A. fumigatus. This difference might be attributed to the geographical distribution of the Aspergillus species complex [13]. This underscores the importance of incorporating local epidemiological data into clinical management practice guidelines, particularly concerning antifungal strategies and treatment outcomes [12].
The clinical implications of present study findings include the necessity for healthcare providers to be more aware of Aspergillus species and their clinical associations, given the susceptibility to fungal resistance and infectious agents within these species. This awareness can facilitate targeted management strategies for affected patients. The initial identification of the Aspergillus species complex was performed using phenotypic methods. However, the accurate identification of cryptic or sibling species of Aspergillus can be challenging and may lead to misidentification using conventional methods [14]. With the development of molecular methods such as sequence-based analysis, precise identification of cryptic species and the description of previously unknown novel species have become possible [15,16]. Based on previous studies, the occurrence of such cryptic species contributes to 10-19% of total Aspergillus infections [16].
The recent rise in the prevalence of cryptic species, along with their repeated isolation from patients with invasive aspergillosis and decreased antifungal susceptibility profiles, necessitates the identification of these rare Aspergillus species [3]. In the present study, several clinically important cryptic species were identified using phylogenetic analysis of four different genes: ITS, hydrophobin, calmodulin, and beta-tubulin genes. The cryptic species identified in this study included A. carneus (n=1) in section Terrei; in section Fumigati-A. lentulus (n=1); A. tamarii (n=2) in section Flavi; and A. tubingensis (n=2), A. welwitschiae (n=2), and A. awamori (n=2) in section Nigri.
Some other clinically important cryptic species reported globally include A. thermomutatus, A. udagawae, A. novofumigatus, A. viridinutans, and A. fumigatiaffinis in the Fumigati section; A. alliaceus in the Flavi section; A. alabamensis in the Terrei section; A. sydowii in the Versicolores section; A. westerdijkiae and A. persii in the Circumdati section; and A. calidoustus and A. keveii in the Usti section [11,12,17,20].
Sequence analysis using ITS, beta-tubulin, and calmodulin has been utilised in previous studies for the accurate identification of cryptic species. In some studies, there is a rare occurrence of discordance in molecular identification between these three genes; the calmodulin gene was preferred for differentiating between species in cases of discrepancies. In the present study, in addition to these three genes, hydrophobin was also used in the sequence analysis of cryptic Aspergillus species, especially in section Fumigati. Besides sequence-based analysis, some automated methods, such as Matrix-Assisted Laser Desorption Ionisation-Time of Flight Mass Spectrometry (MALDI-TOF MS), have also been utilised for identifying cryptic Aspergillus species in various laboratories [18,19].
Due to the reduced susceptibility to routine antifungal drugs observed with these rare cryptic Aspergillus species like A. calidoustus, A. thermomutans, and A. lentulus, there is a strong emphasis on the importance of species-level identification of Aspergillus, especially in patients with invasive aspergillosis [2,22].
Future perspectives should focus on expanding the understanding of the epidemiology and clinical significance of cryptic Aspergillus species, particularly in regions where they may be underreported or overlooked. Additionally, continued advancements in molecular diagnostics, coupled with comprehensive antifungal susceptibility testing, will be essential for improving the management of invasive aspergillosis and other fungal infections.
This study underscores the importance of molecular techniques such as MLSA in accurately identifying Aspergillus species from clinical samples. By providing a detailed characterisation of Aspergillus diversity, present study findings contribute to the existing literature and have implications for clinical practice and future research endeavours.
Limitation(s)
The study has several limitations that warrant consideration. The investigation’s limited generalisability to broader groups may be attributed to two factors: the comparatively small sample size and the fact that it was conducted at a single site. Additionally, the study focused solely on clinical samples from Chennai, South India, potentially overlooking regional variations in Aspergillus species distribution. Furthermore, while MLSA offers precise species-level identification, it is labor-intensive and may not be readily available in all clinical settings. Finally, the retrospective nature of the investigation may introduce bias or result in incomplete data capture, impacting the overall robustness of the results.
Conclusion(s)
Cryptic Aspergillus species are ubiquitous and can be isolated from clinical samples. The highest number of cryptic species was found belonging to the section Nigri. Given the increasing invasive nature of these cryptic species, it is important to perform molecular identification of the isolates for accurate diagnosis of cryptic aspergillosis. Furthermore, conducting antifungal susceptibility testing on these cryptic species would be beneficial to ensure that appropriate antifungal therapy can be initiated.