Introduction
Odontogenic tumours are unusual or unconventional lesions derived from cells associated with odontogenesis and their remnants. These tumours are composed of a heterogeneous group of lesions and are classified based on the odontogenic tissue they mimic: epithelial, mesenchymal, and mixed epithelial-mesenchymal tumours [1]. The pathogenesis of odontogenic tumours is linked to alterations in components of signalling pathways [2]. Various signalling pathways mediate odontogenesis; among them, three major pathways associated with gene mutations include the MAPK pathway, the SHH pathway, and the Wnt signalling pathway. The MAPK pathway is involved in ameloblastoma and Adenomatoid Odontogenic Tumour (AOT), the SHH pathway is implicated in ameloblastoma and Odontogenic Keratocyst (OKC), and the Wnt pathway is associated with odontogenic ghost cell tumours [3,4]. Understanding the core genetic changes in odontogenic tumours could refine their classification, aid in the diagnosis of challenging lesions, and assist in designing new targeted therapies for malignant or aggressive cases. Hence, this review aimed to explain a series of molecular and genetic alterations leading to the progression of these lesions in relation to the MAPK, SHH, and Wnt pathways. Recent advanced technologies, such as Next-Generation Sequencing (NGS), have revealed an increased frequency of gene mutations in odontogenic tumours [5,6].
MAPK/ERK Signalling Pathway
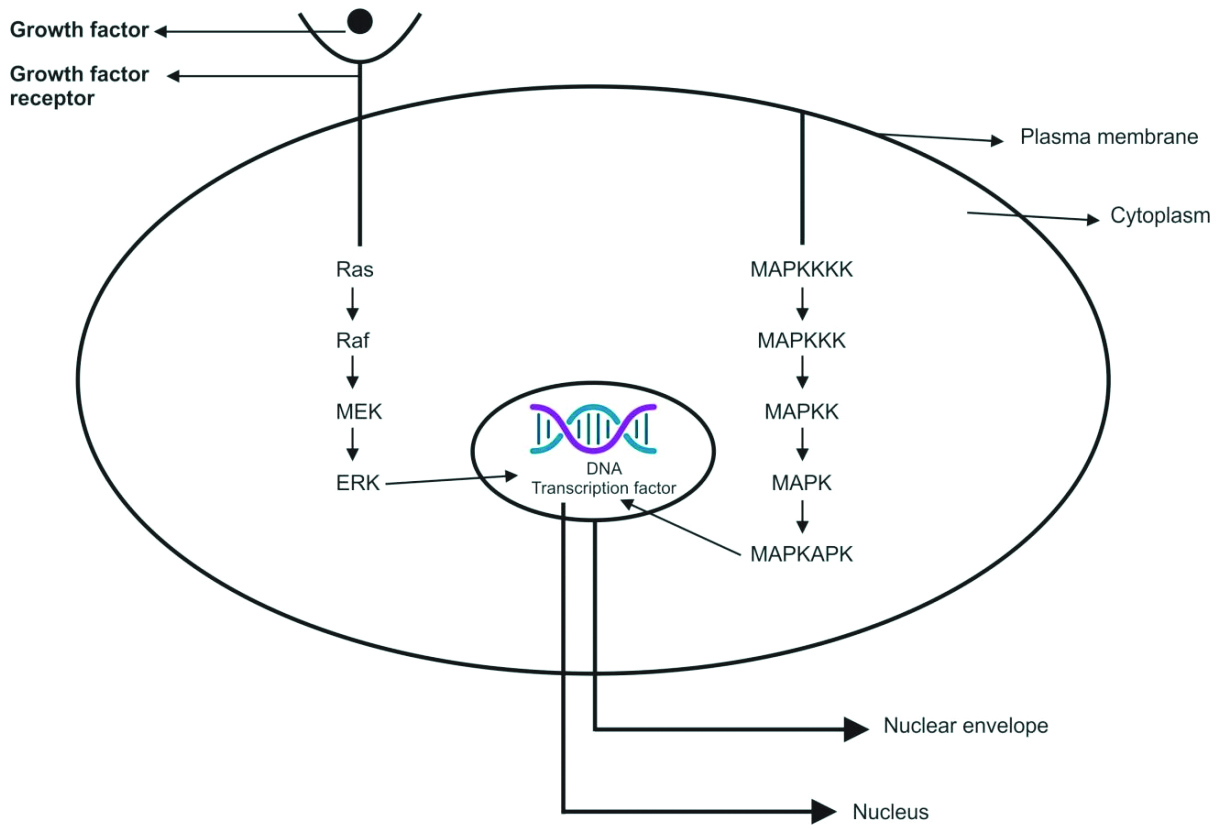
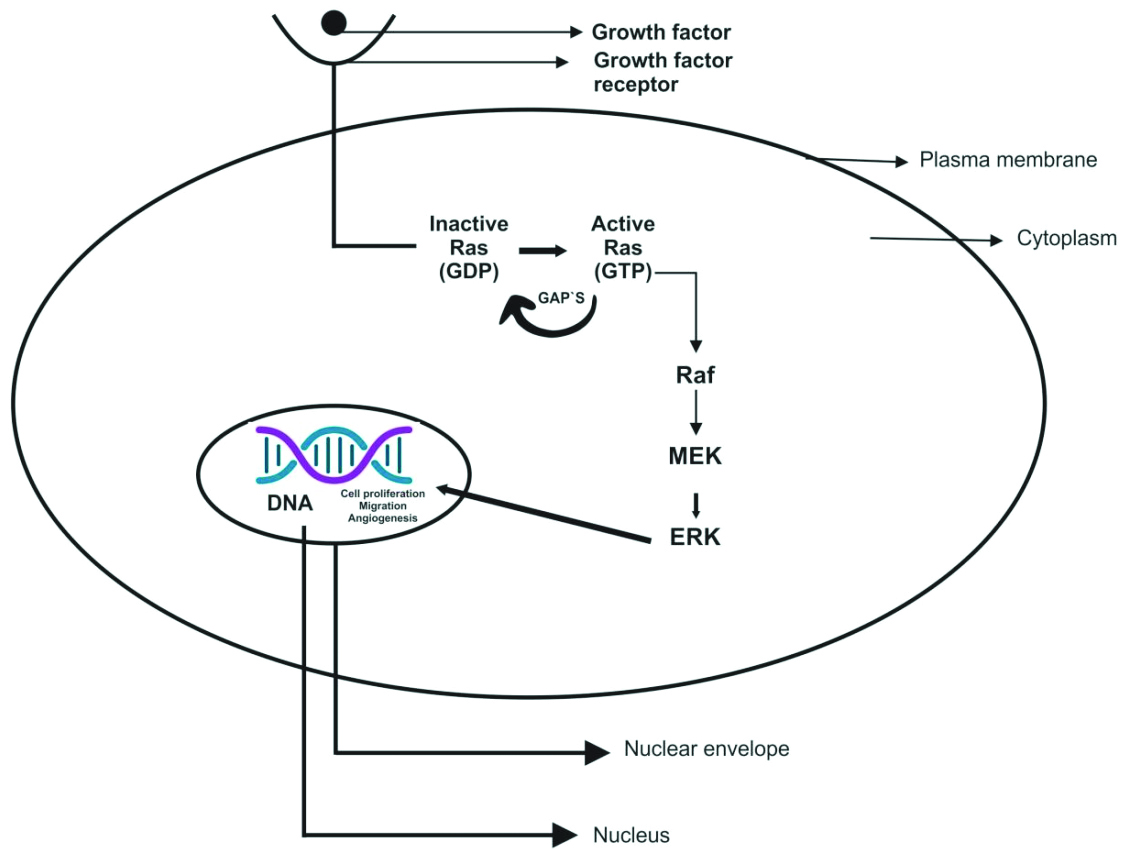
There are various intracellular signalling pathways involved in tumourigenesis, among which the MAPK pathway plays a major role in differentiation, apoptosis, cell proliferation, and tumour metastasis [7]. Extracellular signal-Regulated Kinase (ERK) is a member of the MAPK family [8]. The MAPK signalling pathway consists of three evolutionarily conserved effectors: MAPK Kinase Kinase (MAPKKK), MAPK Kinase (MAPKK), and MAPK [9]. In the RAS-RAF-MEK-ERK cascade of the MAPK/ERK signalling pathway, the intracellular effectors correspond to RAF (MAPKKK), MEK1/2 (MAPKK), and ERK1/2 (MAPK) [Table/Fig-1] [10].
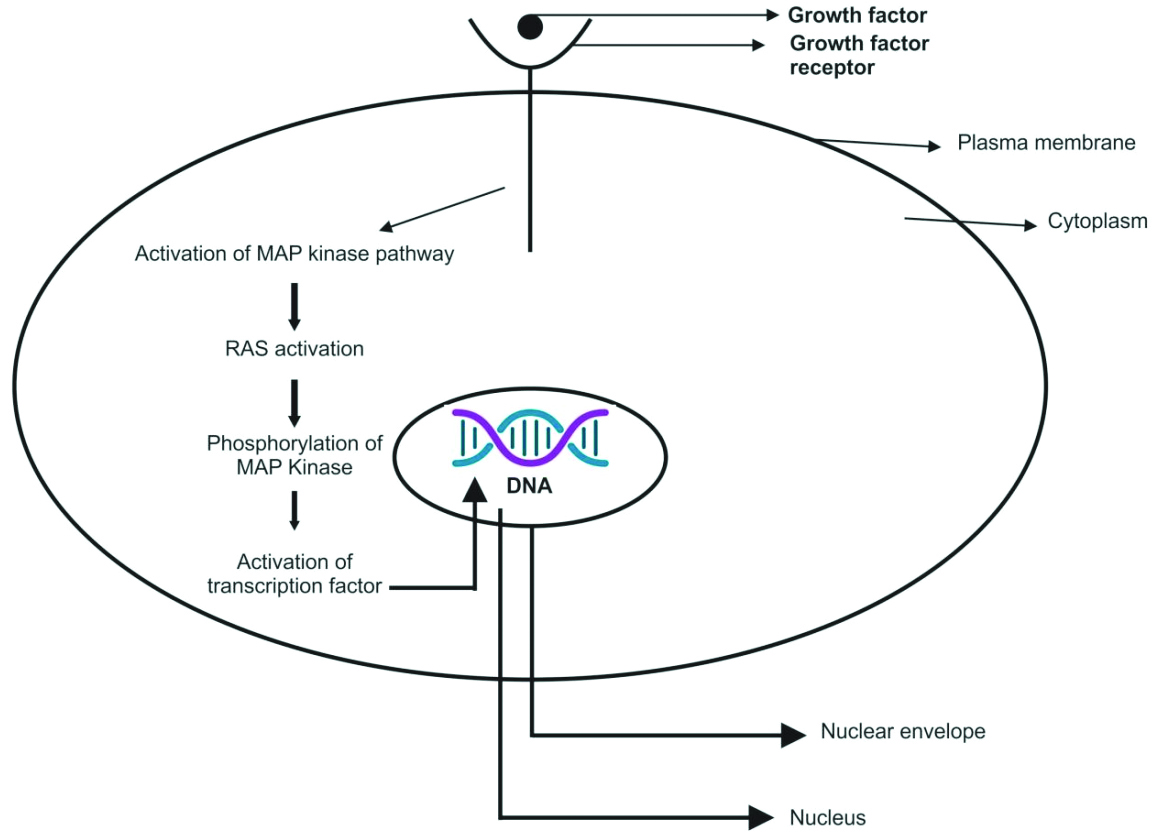
Rat Sarcoma Virus (RAS) is the most commonly mutated oncogene in human tumours. It belongs to a family of small G proteins that bind guanosine nucleotides, specifically Guanosine Triphosphate (GTP) and Guanosine Diphosphate (GDP) [Table/Fig-2] [11]. This protein is located on the inner surface of the cell membrane [12]. Normally, RAS oscillates between an excited, signal-transmitting state and a quiescent state. RAS is inactive when bound to GDP; however, stimulation of the cell by growth factors such as Epidermal Growth Factor (EGF) and Platelet-Derived Growth Factor (PDGF) leads to the exchange of GDP for GTP. This exchange results in a conformational change that generates active RAS. This excited, signal-emitting state is short-lived because of the intrinsic GTPase activity of RAS, which hydrolyses GTP to GDP, releasing a phosphate group and returning the protein to its quiescent, GDP-bound state. The GTPase activity of activated RAS is significantly enhanced by a family of GTPase-Activating Proteins (GAPs), which act as molecular brakes, preventing uncontrolled RAS activation by facilitating the hydrolysis of GTP to GDP. Activated RAS stimulates downstream regulators of proliferation through several interconnected pathways that converge on the nucleus and alter the expression of genes that regulate growth, such as Myelocytomatosis (MYC) [Table/Fig-3] [11].
Overview of cell surface receptors and their principal signal transduction pathway [11].
Action of RAS gene and associated regulators [11].
The RAF protein kinase is composed of 648 Amino Acids (AA) and has a molecular weight ranging from 40-75 kDa. This protein is encoded by the RAF gene [13]. There are three subtypes of the Raf kinase family: 1) A-Raf; 2) B-Raf; and 3) Raf-1. The activation of the Raf kinase pathway occurs through the following mechanisms: i) dimerisation of Raf proteins; ii) localisation of Raf on the inner aspect of the cell membrane through its interaction with RAS; iii) dissociation from Raf kinase inhibitor proteins; iv) phosphorylation and dephosphorylation at different sites; and v) binding to RAS kinase inhibitor [14]. Stokoe D and McCormick F suggested that Raf-1 activation occurs in two steps: A. Ras binding, which fixes Raf-1 on the inner aspect of the cell membrane; B. Activation of Raf-1, which may be conducted by a tyrosine kinase [15].
In the Ras/Raf/MEK/ERK proliferation signal transduction pathway, the activated Ras protein utilises two regions, the Ras binding domain and the cysteine-rich domain at the N-terminal of Raf-1, to bind and translocate Raf from the cytoplasm to the cell membrane, where Raf becomes activated. The activated Raf-1 continues to activate downstream MEK and MAPK, subsequently delivering signals to the nucleus for cell proliferation and differentiation by regulating the activities of transcriptional regulators [Table/Fig-1] [10,16].
MEK (MAPK/ERK kinase): Upon activation of Raf, the C-terminal catalytic domain interacts with MEK, leading to the phosphorylation of its catalytic VIII subregion at a serine residue, thereby activating MEK. The two subtypes of MEK are MEK1 (molecular weight 44 kDa) and MEK2 (molecular weight 45 kDa) [17]. Upon phosphorylation at the tyrosine (Tyr) and threonine (Thr) regulatory sites, MEK activates ERK [18]. ERK (MAPK/ERK) is a serine/threonine (Ser/Thr) protein kinase. Following multiple interactions with protein kinases acting on MEK, the activated MEK interacts directly with ERKs through its N-terminal region, catalysing the phosphorylation of Tyr and Thr residues on ERK to activate it. This signalling stimulates the dimerisation and phosphorylation of ERK, and the activated ERKs translocate to the nucleus to regulate transcription programs, aiding in growth, migration, and differentiation [14,19].
The activation of the MAPK/ERK signalling pathway also activates other extracellular signalling pathways. Extracellular signals such as Vascular Endothelial Growth Factor (VEGF), PDGF and EGF can activate the receptor tyrosine kinase through autologous phosphorylation of the MAPK/ERK signalling pathway. Activated ERK may enter the nucleus, bind to transcription factors, and consequently regulate gene expression, apoptosis, cell proliferation, and differentiation [20].
MAPK (Mitogen-Activated Protein Kinase), MAP4K (MAPK Kinase Kinase Kinase), MAP3K (MAPK Kinase Kinase), MAPKK (MAPK Kinase), MAPKAPK (Mitogen-Activated Protein Kinase-Activated Protein Kinases), and MEK (MAPK/ERK) are key components in cellular signalling pathways [Table/Fig-2] [11].
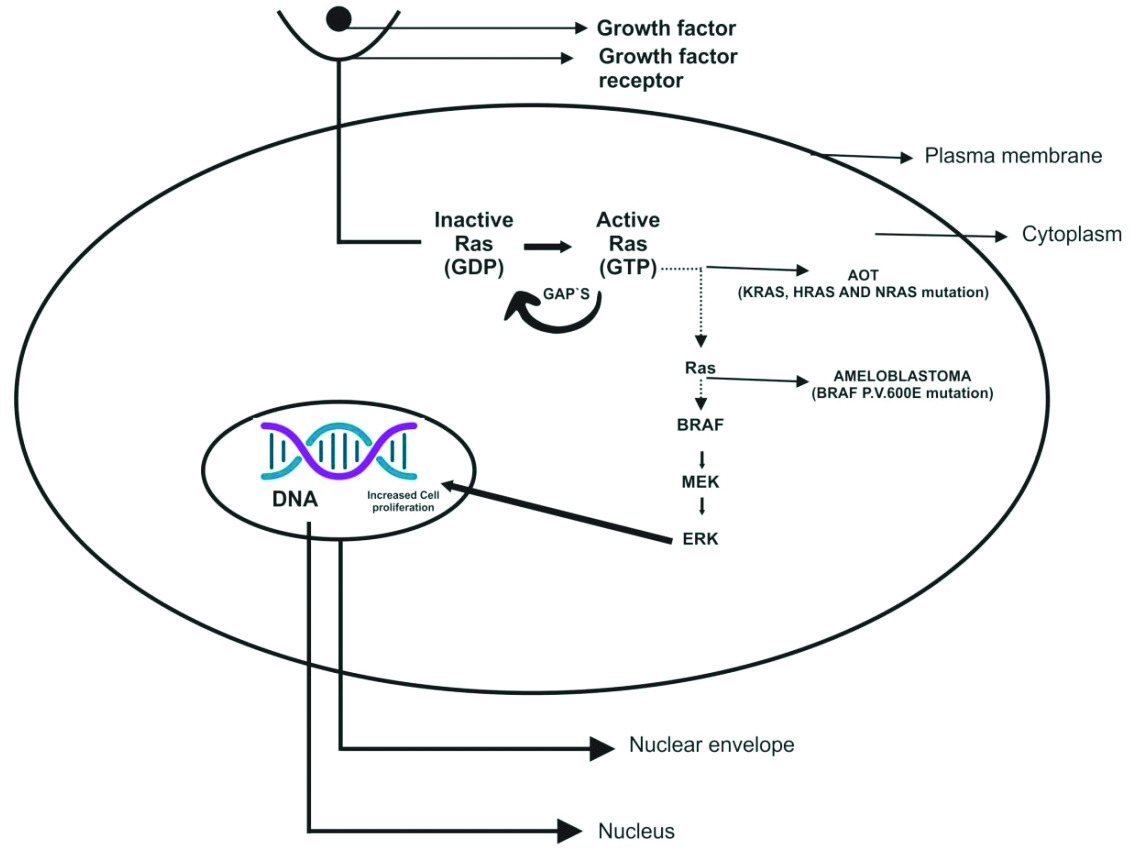
Studies have shown that odontogenic tumours exhibit a high frequency of mutations in the BRAF and RAS genes [Table/Fig-3] [21]. The BRAF mutation results in the substitution of valine (V) to glutamic acid at codon 600 (BRAF P.V600E) and activates the MAPK/ERK signalling pathway. In addition to stimulating this pathway, the BRAF P.V600E mutation promotes cell proliferation [22]. The BRAF P.V600E mutation is commonly observed in ameloblastoma. The RAS proto-oncogene family, which includes KRAS, NRAS, and HRAS, plays a crucial role in cancer. These genes encode RAS GTPases, which act as upstream activators of MAPK/ERK signalling by activating RAF proteins [23]. RAS mutations also activate the MAPK/ERK pathway. A study by Coura BP et al., identified mutations in the KRAS gene (p.G12V and p.G12R) and the BRAF P.V600E gene in the normal odontogenic apparatus [24].
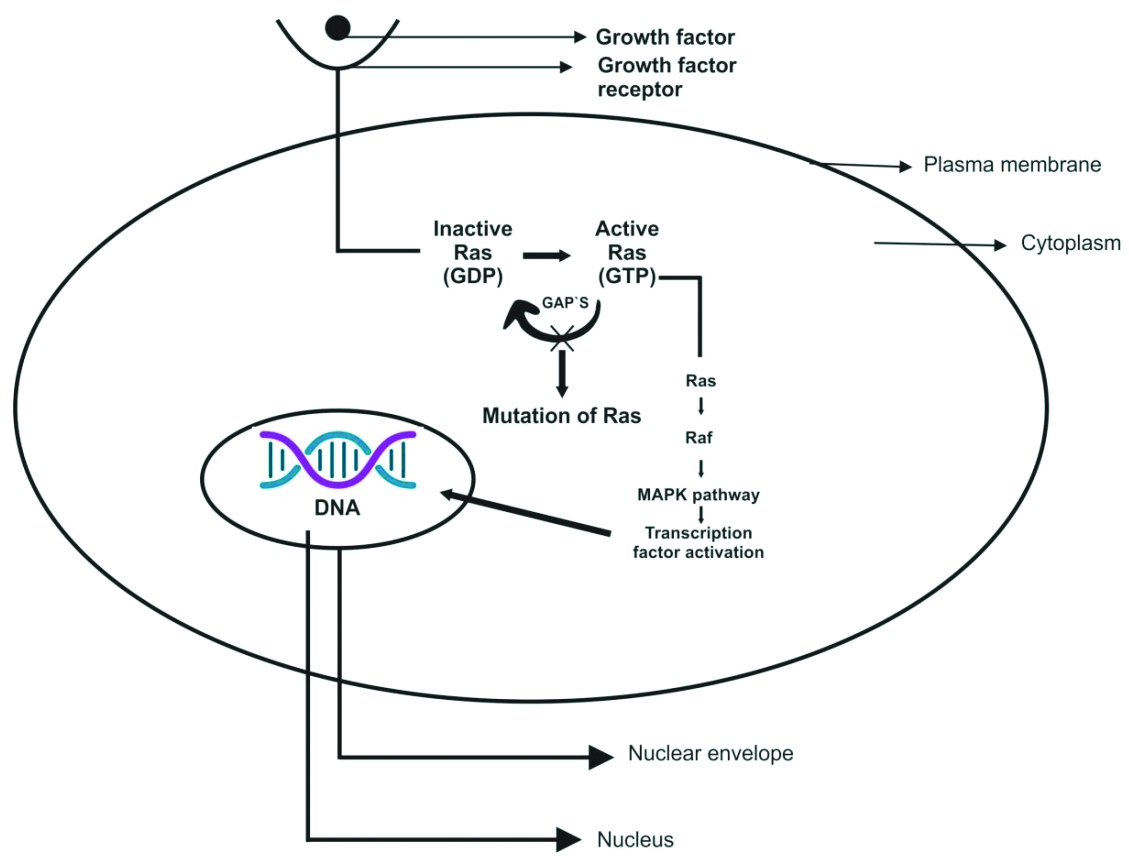
The MAPK/ERK signalling pathway operates under normal circumstances and in the presence of activating mutations, as illustrated in [Table/Fig-4] [25]. (a) The canonical RAS-RAF-MEK-ERK cascade is activated by external signals (e.g., EGF growth factors) binding to receptor tyrosine kinases, such as Fibroblast Growth Factor Receptor (FGFR) and Epidermal Growth Factor Receptor (EGFR). This interaction triggers signalling through RAS-GTP, RAF, MEK, and ERK, culminating in the action of phosphorylated ERK on its substrates and the regulation of various cellular biological functions. In the absence of external stimuli, a switch occurs between the active (RAS-GTP) and inactive (RAS-GDP) forms due to the action of GAPs, which promote hydrolysis. (b) In the presence of the BRAF P.V600E mutation, MAPK/ERK signalling is progressively activated by BRAF, even in the absence of growth factors and dimerisation with RAS, thereby supporting continuous MAPK/ERK signalling. (c) The imbalance between the inactive and active RAS forms is caused by mutations in the RAS genes (KRAS, NRAS, and HRAS), which lead to a promotion of the active state (RAS-GTP) by either decreasing GTP hydrolysis or increasing the rate of GTP loading. This mechanism sustains MAPK/ERK signalling constitutively, even without external stimuli [25].
MAPK/ERK signalling pathway under normal circumstances and in the presence of activating mutations [25].
The functions of the MAPK/ERK signalling pathway include cell proliferation, cell differentiation, cell cycle regulation, apoptosis, and tumour formation. Specifically:
i) The MAPK/ERK signalling pathway promotes cell proliferation [Table/Fig-5] and has antiapoptotic effects; thus, inhibition of this pathway reduces cell proliferation and antiapoptotic responses [26].
ii) It promotes tumour invasion and apoptosis; initially, in tumour cells, loss of cohesion results in the destruction of the basement membrane, leading to invasion [27].
iii) It is involved in the degradation of the Extracellular Matrix (ECM) by Matrix Metalloproteinases (MMPs), facilitating the invasion and metastasis of cancer cells [28].
iv) It promotes tumour angiogenesis by expressing IL-8 and VEGF in various tumours [29].
Role of MAPK/ERK signalling pathway [11].
Clinical implications: It was observed that among all the histological subtypes of ameloblastoma, the plexiform pattern has a higher frequency of SMO mutations, while follicular ameloblastoma is more commonly associated with BRAF V600E mutations. In immunohistochemical testing, ameloblastic lesions such as peripheral ameloblastoma, unicystic ameloblastoma, ameloblastic fibroma, and ameloblastic fibroodontoma have demonstrated BRAF V600E positivity. Ameloblastoma of the type AOT is a benign odontogenic tumour characterised by spindle-shaped epithelial cells arranged in rosettes and duct-like structures. AOT also exhibits mutations in the MAPK pathway.
Hedgehog Pathway
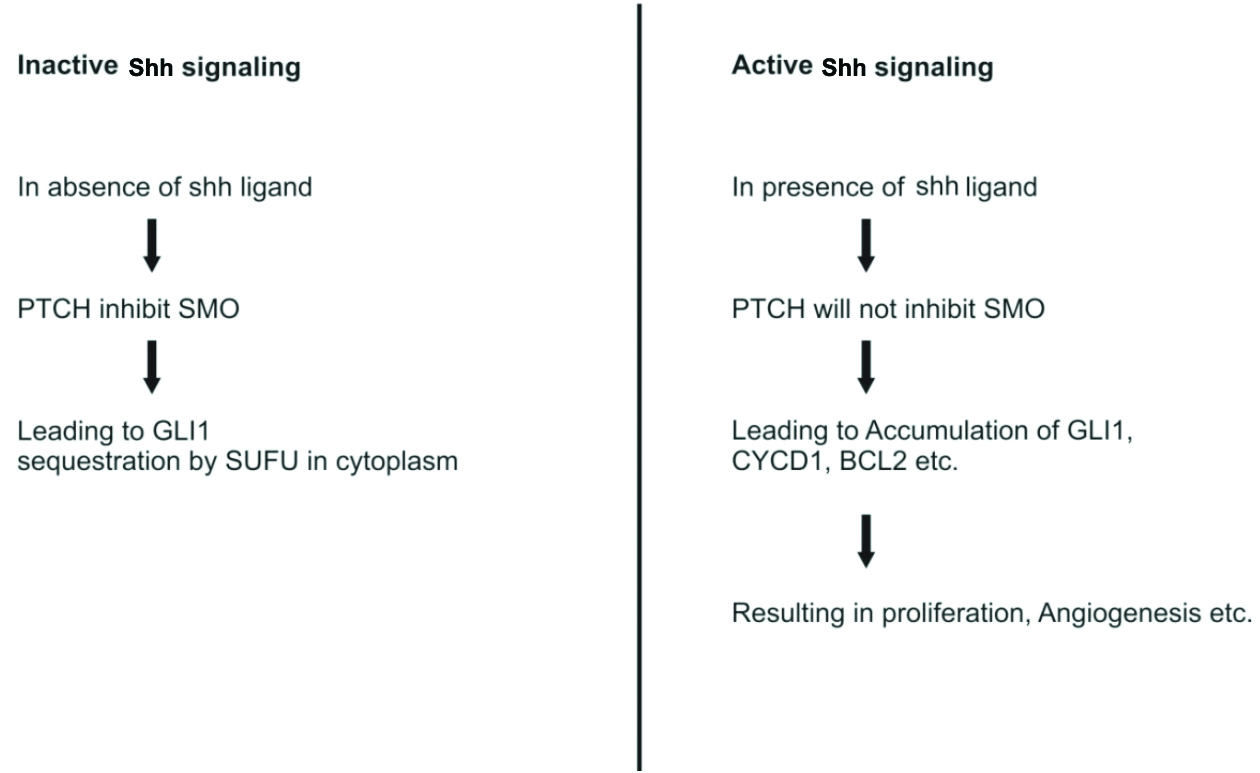
As a component of the Hedgehog (Hh) family, Sonic Hedgehog (Shh) was originally identified in Drosophila melanogaster, and the Shh signalling pathway is highly expressed in mammals [30]. Upon binding to its receptor protein, Patched Tumour Suppressor gene (PTCH), Shh can increase the activity of Smoothened (SMO) and Glioma-associated oncogene homolog (GLI) transcription factors, subsequently controlling the expression of target genes that regulate cell growth and survival in several tissues. At the beginning of the Hh signalling pathway, the binding of the Hh ligand occurs. In the absence of the ligand, PTCH1 represses (controls) SMO. When the ligand binds, it relieves this inhibition, allowing SMO to modulate Suppressor of Fused Homolog (SUFU), which in turn modifies GLI1 (glioma-associated oncogene homolog 1) [Table/Fig-6] [31].
Active and inactive- Hedgehog signalling pathway [31].
PTCH: Patched tumour suppressor gene; SMO: Smoothened; SUFU: Suppressor of fused homolog; GLI1: Glioma-associated oncogene homolog 1
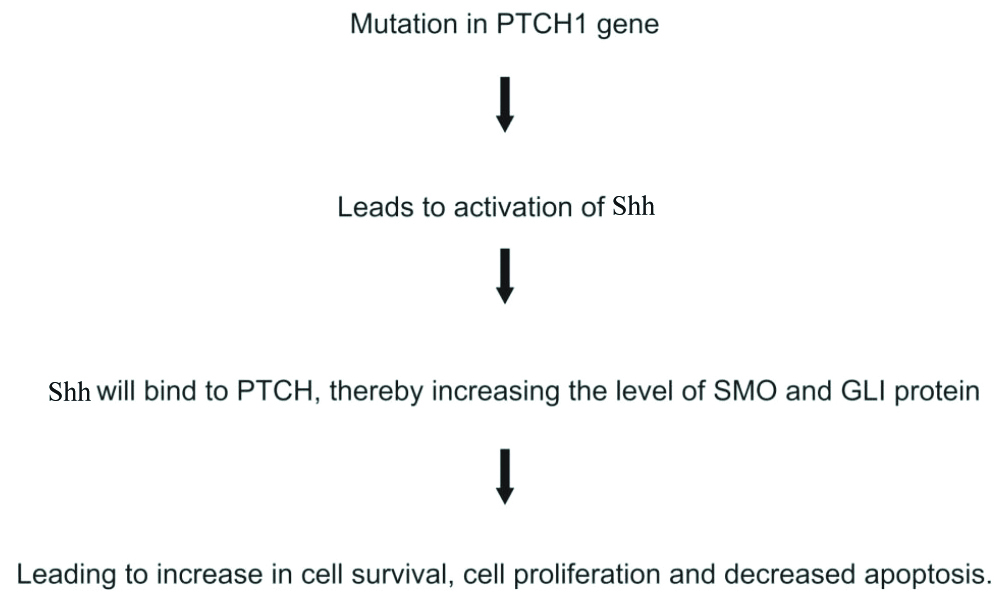
Activation of the Shh pathway due to PTCH1 mutation is considered the most important mechanism involved in the pathogenesis of Odontogenic Keratocyst (OKC) [32]. [Table/Fig-7] shows the role of PTCH1 mutation in the pathogenesis of odontogenic tumours such as OKC and ameloblastoma. Shh increases the production of Bcl-2, which results in apoptotic resistance and an increase in cell growth [33]. In OKC, increased Bcl-2 positivity has been reported previously [34]. SOX2 is a transcription factor that enhances the proliferative capacity of cells and promotes survival. Its production is regulated by Shh [35]. The overexpression of SOX2 in the basal and suprabasal layers of OKC has been reported by Bandyopadhyay A et al., [36]. In ameloblastoma, the expression of Shh, PTCH, SMO, and Gli1 mRNA has been observed [37].
Role of PTCH1 mutation in pathogenesis of odontogenic lesions such as Ameloblastoma (AB) and OKC [32].
Clinical implications: PTCH1 is found to be mutated in 85% of syndromic KCOT/OKC cases (in association with nevoid basal cell carcinoma). Other Shh pathway mutations, such as SUFU and PTCH2, are seen in patients with nevoid basal cell carcinoma syndrome at a lower rate.
WNT Signalling Pathway
The name Wnt is derived from the fusion of the names of the Drosophila segment polarity gene “wingless” and the vertebrate homologue, integrated or int-1 [38]. Wnt proteins constitute a large family of glycoproteins that are secreted and play crucial roles in various biological processes such as embryonic development, cell differentiation, cell motility, cell proliferation, and maintenance of tissue equilibrium [39].
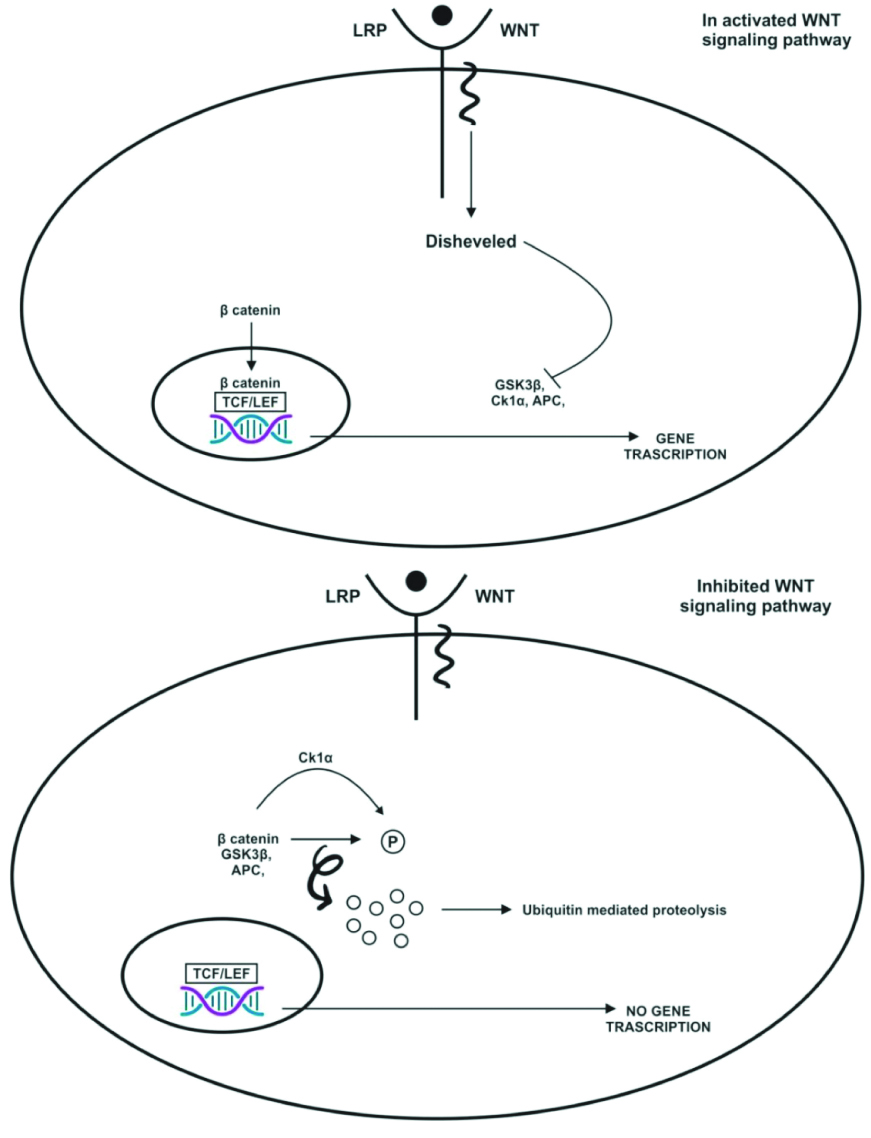
In the activated Wnt signalling pathway, binding of the Wnt ligand to the Frizzled (FZD) receptor and Lipoprotein Receptor-related Protein (LRP) co-receptor occurs. The LRP receptors are phosphorylated by CK1α and GSK3β, which recruit Dishevelled (DVL) protein. These proteins are polymerised and activated. DVL inactivates the destructive complex, resulting in the stabilisation and accumulation of beta-catenin. This beta-catenin translocates to the nucleus and forms an active complex with T-cell Factor/Lymphoid Enhancer Factor (TCF/LEF). This transcriptional switch leads to changes in multicellular processes.
In the inhibited Wnt signalling pathway, the absence of Wnt ligands leads to the phosphorylation of beta-catenin by the destructive complex (which contains APC, AXIN, CK1α, and GSK3β). Beta-catenin is phosphorylated by GSK3β, which targets it for proteasomal degradation. In the absence of beta-catenin, a repressive complex containing TCF/LEF recruits to suppress target genes [Table/Fig-8] [39].
Wnt pathway activated and inhibited [39].
In the downregulation of the Wnt signalling pathway for the regulation of beta-catenin degradation, the APC gene is involved [40]. It regulates the cell cycle, cell migration, and cell division [41]. In Calcifying Cystic Odontogenic Tumours (CEOT), mutations in beta-catenin have been observed [42]. [Table/Fig-9] shows the Wnt/beta-catenin pathway in normal cells and odontogenic lesions [32].
Role of WNT-β catenin pathway [32].

Clinical implication: The family of odontogenic ghost cell tumours is composed of the CEOT, which was reclassified in 2017 as the Calcifying Odontogenic Cyst (COC), as well as the Dentinogenic Ghost Cell Tumour (DGCT) and Odontogenic Ghost Cell Carcinoma (OGCC). CEOT accounts for 93% of odontogenic ghost cell tumours, while DGCT and OGCC represent 5% and 2%, respectively. Tumours in this group (i.e., odontogenic ghost cell tumours) are associated with mutations in the CTNNB1 (β-catenin) gene within the Wnt signalling pathway.
Conclusion(s)
This review summarises how the ERK/MAPK, Shh, and Wnt signalling pathways play a role in the development of tumours. Genomic and proteomic profiling are needed to clarify the pathogenesis of odontogenic tumours. The development and progression of odontogenic lesions depend on a wide variety of factors. A deeper understanding of the pathogenesis of odontogenic lesions assists in determining patient prognosis and designing more targeted therapeutic treatments, ultimately decreasing the mortality and morbidity of patients.
[1]. El-Naggar AK, Chan John KC, Grandis JR, Takata T, Slootweg PJ, World Health Organization Classification of Head and Neck Tumours 2017 4th edLyon, FranceWorld Health Organization:347 [Google Scholar]
[2]. Diniz MG, Gomes CC, de Sousa SF, Xavier GM, Gomez RS, Oncogenic signalling pathways in benign odontogenic cysts and tumoursOral Oncol 2017 72:165-73. [Google Scholar]
[3]. Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, Activating FGFR2-RAS-BRAF mutations in ameloblastomaClin Cancer Res 2014 20(21):5517-26. [Google Scholar]
[4]. Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, Identification of recurrent SMO and BRAF mutations in ameloblastomasNat Genet 2014 46(7):722-25. [Google Scholar]
[5]. Guimarães LM, Coura BP, Gomez RS, Gomes CC, The molecular pathology of odontogenic tumours: Expanding the spectrum of MAPK pathway driven tumoursFront Oral Health 2021 14(2):740-88. [Google Scholar]
[6]. Koboldt DC, Steinberg KM, Larson DE, Wilson RK, Mardis ER, The next-generation sequencing revolution and its impact on genomicsCell 2013 155(1):27-38. [Google Scholar]
[7]. Wortzel I, Seger R, The ERK cascade: Distinct functions within various subcellular organellesGenes Cancer 2011 2(3):195-209. [Google Scholar]
[8]. Yang S, Liu G, Targeting the Ras/Raf/MEK/ERK pathway in hepatocellular carcinomaOncol Lett 2017 13(3):1041-47. [Google Scholar]
[9]. Krishna M, Narang H, The complexity of mitogen-activated protein kinases (MAPKs) made simpleCell Mol Life Sci 2008 65:3525-44. [Google Scholar]
[10]. Guo Y, Pan WW, Liu SB, Shen ZF, Xu Y, Hu L, ERK/MAPK signalling pathway and tumourigenesis (Review)Exp Ther Med 2020 19(3):1997-2007. [Google Scholar]
[11]. Kumar V, Cotran R, Robbins S, Robbins Basic Pathology 2005 7th edElesvier [Google Scholar]
[12]. Yoon S, Seger R, The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functionsGrowth Factors 2006 24(1):21-44. [Google Scholar]
[13]. Terrell EM, Morrison DK, Ras-Mediated Activation of the Raf Family KinasesCold Spring Harb Perspect Med 2019 9(1):a033746 [Google Scholar]
[14]. Roskoski R, Targeting ERK1/2 protein-serine/threonine kinases in human cancersPharmacol Res 2019 142:151-68. [Google Scholar]
[15]. Stokoe D, McCormick F, Activation of c-Raf-1 by Ras and Src through different mechanisms: Activation in vivo and in vitroEMBO J 1997 16(9):2384-96. [Google Scholar]
[16]. Lavoie H, Gagnon J, Therrien M, ERK signalling: A master regulator of cell behaviour, life and fateNat Ver Mol Cell Biol 2020 21(10):607-32. [Google Scholar]
[17]. Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S, The protein kinase complement of the human genomeScience 2002 298(5600):1912-34. [Google Scholar]
[18]. Muta Y, Matsuda M, Imajo M, Divergent dynamics and functions of ERK MAP kinase signaling in development, homeostasis and cancer: Lessons from fluorescent bioimagingCancers (Basel) 2019 11(4):513 [Google Scholar]
[19]. Maik-Rachline G, Hacohen-Lev-Ran A, Seger R, Nuclear ERK: Mechanism of translocation, substrates, and role in cancerInt J Mol Sci 2019 20(5):1194 [Google Scholar]
[20]. Kolch W, Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactionsBiochem J 2000 351(Pt 2):289-305. [Google Scholar]
[21]. Coura BP, Bernardes VF, de Sousa SF, França JA, Pereira NB, Pontes HAR, KRAS mutations drive adenomatoid odontogenic tumour and are independent of clinicopathological featuresMod Pathol 2019 32(6):799-806. [Google Scholar]
[22]. Dhillon AS, Hagan S, Rath O, Kolch W, MAP kinase signalling pathways in cancerOncogene 2007 26(22):3279-90. [Google Scholar]
[23]. Haigis KM, KRAS Alleles: The devil is in the detail trendsCancer 2017 3(10):686-97. [Google Scholar]
[24]. Coura BP, de Resende TAC, de Menezes VCB, Bernardes VF, de Sousa SF, Diniz MG, Assessing pathogenic mutations in dental follicles as an attempt to identify early events in odontogenic tumours tumourigenesisArch Oral Biol 2020 113:104523 [Google Scholar]
[25]. Eblen ST, Extracellular-regulated kinases: Signaling from ras to ERK substrates to control biological outcomesAdv Cancer Res 2018 138:99-142. [Google Scholar]
[26]. Baek JH, Jang JE, Kang CM, Chung HY, Kim ND, Kim KW, Hypoxia-induced VEGF enhances tumoursurviv- ability via suppression of serum deprivation-induced apoptosisOncogene 2000 19(40):4621-31. [Google Scholar]
[27]. Sulzmaier FJ, Ramos JW, RSK isoforms in cancer cell invasion and metastasisCancer Res 2013 73(20):6099-105. [Google Scholar]
[28]. Gialeli C, Theocharis AD, Karamanos NK, Roles of matrix metalloproteinases in cancer progression and their pharmaco-logical targetingFEBS J 2011 278(1):16-27. [Google Scholar]
[29]. Soula-Rothhut M, Coissard C, Sartelet H, Boudot C, Bellon G, Martiny L, The tumour suppressor PTEN inhibits EGF-induced TSP-1 and TIMP-1 expression in FTC-133 thyroid carcinoma cellsExp Cell Res 2005 304(1):187-201. [Google Scholar]
[30]. Fietz MJ, Concordet JP, Barbosa R, Johnson R, Krauss S, McMahon AP, The hedgehog gene family in drosophila and vertebrate developmentDev Suppl 1994 :43-51. [Google Scholar]
[31]. Lee DH, Lee SY, Oh SC, Hedgehog signaling pathway as a potential target in the treatment of advanced gastric cancerTumour Biol 2017 39(6):1010428317692266 [Google Scholar]
[32]. Bhuyan L, Nishat R, Behura SS, Mahapatra N, Kumar H, Insight into the molecular pathogenesis of odontogenic lesionsJ Oral Biosci 2021 63(1):35-44. [Google Scholar]
[33]. Lee RTH, Zhao Z, Ingham PW, Hedgehog signallingDevelopment 2016 143:367-72. [Google Scholar]
[34]. Razavi SM, Torabinia N, Mohajeri MR, Shahriyary S, Ghalegolab S, Nouri S, Expression of Bcl-2 and epithelial growth factor receptor proteins in keratocysticod ontogenic tumour in comparison with dentigerous cyst and ameloblastomaDent Res J 2015 12(4):342-47. [Google Scholar]
[35]. Santini R, Pietrobono S, Pandolfi S, Montagnani V, D’Amico M, Penachioni JY, SOX 2 regulates self-renewal and tumourigenicity of human melanoma-initiating cellsOncogene 2014 33(38):4697-708. [Google Scholar]
[36]. Bandyopadhyay A, Nishat R, Behura SS, Panda A, Ramachandra S, Mohiddin G, Cancer stem cell markers, SOX 2 and OCT 4 in ameloblastoma and keratocystic odontogenic tumour: An immunohistochemical studyJ Int Oral Health 2017 9(1):28-32. [Google Scholar]
[37]. Gurgel CA, Buim ME, Carvalho KC, Sales CB, Reis MG, de Souza RO, Transcriptional profiles of SHH pathway genes in keratocystic odontogenic tumour and ameloblastomaJ Oral Pathol Med 2014 43(8):619-26. [Google Scholar]
[38]. Zhang Y, Wang X, Targeting the Wnt/β-catenin signaling pathway in cancerJ Hematol Oncol 2020 13(1):165 [Google Scholar]
[39]. Liu J, Xiao Q, Xiao J, Niu C, Li Y, Zhang X, Wnt/β-catenin signalling: Function, biological mechanisms, and therapeutic opportunitiesSignal Transduct Target Ther 2022 7(1):3 [Google Scholar]
[40]. Nishat R, Behura SS, Ramachandra S, Kumar H, Bandyopadhyay A, Human Papilloma Virus (HPV) induced head & neck squamous cell carcinoma: A comprehensive retrospectJ Clin Diagn Res 2015 9(6):ZE01-ZE04. [Google Scholar]
[41]. Li N, Liu B, Sui C, Jiang Y, Analysis of APC mutation in human ameloblastoma and clinical significanceSpringer Plus 2016 5:314 [Google Scholar]
[42]. Dutra SN, Pires FR, Armada L, Azevedo RS, Immunoexpression of Wnt/β-catenin signaling pathway proteins in ameloblastoma and calcifying cystic odontogenic tumorJ Clin Exp Dent 2017 9(1):e136-e140. [Google Scholar]