Introduction
Alzheimer’s Disease (AD) is a chronic progressive neurodegenerative disorder characterised by declining memory, personality changes, brain atrophy, and synaptic dysfunctions [1]. Epidemiological evidence shows that approximately 57.4 million people are currently affected by dementia, and this number is predicted to surpass 152.8 million by 2050, making it one of the major health burdens globally [2]. The aggregation of β-amyloid (Aβ) plaques and Neurofibrillary Tangles (NFT) are well known key pathological hallmarks of AD [3]. The accumulation of β-amyloid in the brain is implicated as the primary trigger of the pathomechanism of AD. However, the pathogenesis of Aβ formation in the cerebral cortex (temporal lobe) is not yet fully understood. In this context, present review aimed to determine the pathomechanism of the protein (Aβ) in the formation of amyloid plaques and its interaction with other biological markers that could aggravate the disease condition.
Risk Factors
AD is a multifactorial disease that develops from a plethora of known contributing factors, such as ageing, genetic predisposition, and family history. Among these, ageing is one of the most common risk factors. The total number of people with AD dementia is projected to reach 13.8 million by 2050, with 7.0 million individuals aged 85 years or older [4]. Additionally, the APOE4 genotype has a significant impact on increasing Aβ deposition, leading to Late-Onset Alzheimer’s Disease (LOAD). Individuals with a family history of Alzheimer’s-specifically those who have or had a parent or sibling (first-degree relative) with the disease-are more likely to develop the condition than those without a first-degree relative affected by AD [5].
Genetic factors: The majority of AD cases are sporadic and induced by several pathological markers, while early-onset AD (EOAD) may result from autosomal dominant mutations in certain genes, including the Amyloid Precursor Protein (APP), presenilin-1 (PS-1), and presenilin-2 (PS-2). One of the allelic variants of the apolipoprotein E gene, APOE4, is considered the strongest genetic factor for the initiation of both EOAD and LOAD [6].
Vascular risk factors: Recent literature highlights modifiable risk factors such as hypertension, diabetes, obesity, increased alcohol intake, smoking, and Cerebrovascular Accidents (CVA) that increase the likelihood of developing the pathophysiology of AD or other forms of Alzheimer’s-Related Dementia (ARD) [6,7].
Psychosocial factors: Psychosocial factors are also known to contribute to the development of cognitive impairment and dementia. Several studies have identified loneliness, depression, sleep disturbances, anxiety, hallucinations, and low Socio-economic Status (SES) as important contributing factors in the pathogenesis of AD [8].
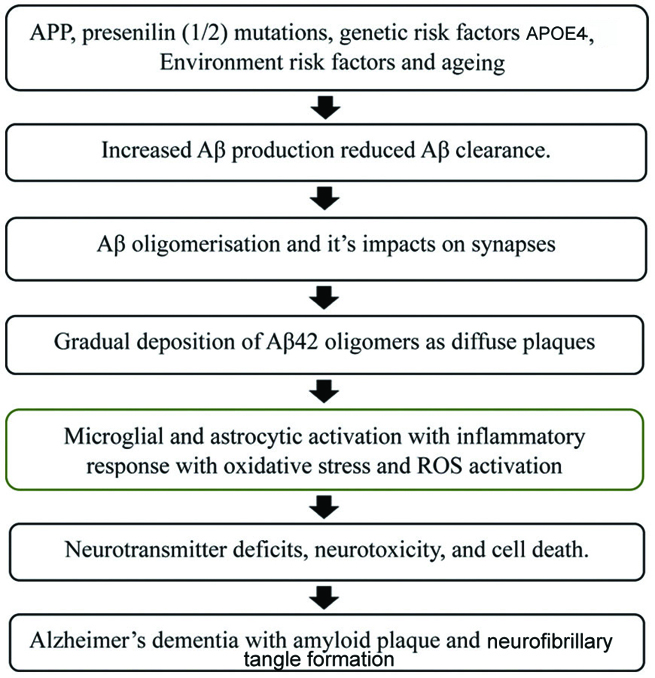
Amyloid cascade hypothesis: In 1992, Hardy JA and Higgins GA proposed the amyloid cascade hypothesis for the pathophysiology of AD [Table/Fig-1] [9]. This hypothesis illustrates that the aggregation of fibrillary Aβ is the principal contributing factor in the development of AD. Furthermore, the prominent occurrence of Aβ plaques and mutations within the Amyloid Precursor Protein (APP) pathway affect the generation of Aβ plaques, which leads to Familial Alzheimer’s Disease (FAD). Amyloid plaques are insoluble protein clumps formed by the cleavage of APP by the γ-secretase complex, as well as by the production of 42-amino acid peptides known as Aβ-42 or Aβ [10]. These amyloid plaques exacerbate the pathogenesis of AD dementia and cognitive impairment, a phenomenon collectively termed the “amyloid cascade hypothesis.” Additionally, the aggregation of beta-amyloid is considered a intrinsic component in the formation of NFT, vascular damage, and cellular death. The cleavage of APP by beta-site APP-cleaving enzyme (BACE) and gamma-secretase involves a 695-amino acid membrane protein. The cleavage sites of APP by these enzymes differ at the N-terminal domain of the Aβ sequence, and gamma-secretase initiates the action of presenilins (1 and 2), which cleave APP through endopeptidase and carboxylase activities. Consequently, mutations in APP and presenilins (1 and 2) contribute equally to FAD [11]. However, other mechanisms, including ageing, oxidative stress, and genetic mutations, can disrupt Aβ homeostasis, potentially leading to the accumulation of plaques and the production of Aβ oligomers. These Aβ oligomers are significantly neurotoxic and result in neuronal death, cognitive impairment, and dementia [12].
Amyloid cascade hypothesis explains the pathogenesis of Alzheimer’s Disease (AD) [9].
APP: Amyloid precursor protein; APOE: Apolipoprotein E gene; Aβ: Amyloid beta; ROS: Reactive oxygen species
Formation of Aβ oligomers and fibrils: The transformation of Aβ from a monomeric to a fibrillar form during its molecular life cycle passes through an oligomeric form (Aβos). The unfolded monomers, upon joining with partially folded monomers, form transient small Aβ oligomers. Once structured Aβ oligomers are formed, protofibrils (β-sheets) are added to create fibrils, which aggregate to form amyloid plaques [13]. Cellular degradation and clearance pathways assist in removing misfolded proteins and preventing the aggregation of fibrils. Thus, any disturbances in these pathways result in the deposition of amyloid proteins [14]. During protein aggregation, secondary nucleation on Aβ amyloid fibrils produces Aβ-42 oligomers, which are associated with the process of amyloid genesis [15]. The interaction of monomeric Aβ with the α1β1 complex of Na-K ATPase disrupts neuronal transmembrane potential, while the association between adrenergic receptor α2A and Aβ oligomers activates glycogen synthase kinase 3β (GSK-3β) and induces hyperphosphorylation of tau protein. Additionally, the toxicity of Aβ oligomers substantially causes overactivation of N-methyl-D-aspartate receptors (NMDAR), resulting in direct neuronal injury and promoting cell death [16].
Aβ biogenesis: Aβ is derived from the breakdown of APP by BACE1 and γ-secretase, while APP is encoded by the APP gene on chromosome 21 [17]. However, α-secretase processes APP through a non toxic mechanism to generate Aβ. The intracellular Aβ is distributed in the neuronal cytosol and the Endoplasmic Reticulum (ER), which are normal sites for its regulation. BACE1 cleaves APP at the Asp1 or Glu11 positions of the Aβ sequence, while γ-secretase cleaves presenilin at the ε-, ς-, and γ-cleavage sites, releasing the Aβ Intracellular Domain (AICD). Additionally, abnormal mutations in presenilin due to environmental factors can disrupt γ-secretase activity, potentially leading to smaller cuts in the APP molecule and ultimately resulting in a longer Aβ peptide chain [18]. The monomeric form of Aβ clusters to form toxic oligomers, which aggregate into protofibrils and eventually into fibrils containing β-sheets. Thus, increased production of Aβ is a well known contributing factor to the development of AD dementia [19].
Toxicity of Aβ: The formation of Aβ plaques is a key factor that induces neurotoxicity and cell death. Furthermore, Aβ interacts with specific cell surface receptors and proteins, triggering the production of Reactive Oxygen Species (ROS), which causes cytotoxic effects and alterations in the nervous system, as well as mitochondrial dysfunction. Additionally, ROS react with proteins and lipids to form oxidised proteins and oxidised lipids, known as lipid peroxides, which can disrupt biological structures, including membrane integrity, and potentially affect crucial enzymes such as Glutamine Synthetase (GS) and Creatine Kinase (CK), both of which are vital for neuronal function [20,21]. Moreover, lipid peroxidation (LPO) generates toxic products like 4-Hydroxy-2-Nonenal (HNE) and 2-propenal (acrolein), which exert hazardous effects on neuronal structures, cellular function, Ca2+ metabolism, and mitochondrial dysfunction, contributing to neurotoxicity. Furthermore, Aβ plaques mediate the release of inflammatory chemicals, including eicosanoids, chemokines, and proinflammatory cytokines, which hinder the microglial clearance of Aβ. As a result, this leads to microglial-mediated apoptosis, loss of synaptic plasticity, inflammation, and excitotoxicity, all of which are associated with neurotransmitter receptors and ultimately contribute to AD. Association of Aβ with other biochemical markers in the pathogenesis of Alzheimer’s disease [Table/Fig-2].
Association of Amyloid Beta (Aβ) with other principal biomarkers.
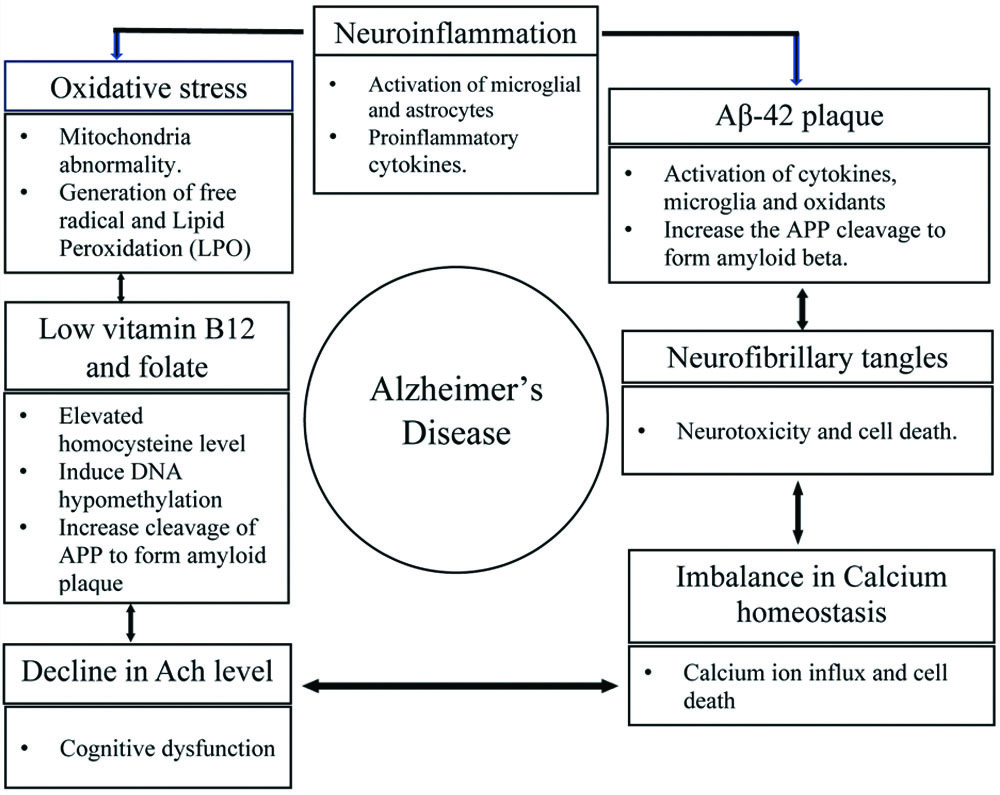
Elevated homocysteine levels: Raised homocysteine levels, along with decreased vitamin B12 and folate, are suggestive factors in the development of neurodegenerative disorders, including AD. Homocysteine is a key component of methionine metabolism, which includes its remethylation by Methionine Synthase (MS), influenced by vitamin B12 and folate as intrinsic cofactors, while 5-methyl tetrahydrofolate (THF) serves as the methyl donor. Furthermore, methionine interacts with Adenosine Triphosphate (ATP) to form S-Adenosyl Methionine (SAM), catalysed by the enzyme S-adenosylmethionine synthase, which utilises the active form of vitamin B6, pyridoxal-5’-phosphate (PLP), as a coenzyme. Later in the metabolic cycle, depletion of SAM results in the formation of S-adenosylhomocysteine (SAH), which is subsequently hydrolysed into homocysteine. Moreover, inborn errors in vitamin B12 and folic acid metabolism affect the regulation of the SAM/SAH ratio, leading to elevated homocysteine levels (hyperhomocysteinemia). Hyperhomocysteinemia exacerbates DNA hypomethylation, which may induce mutations in the presenilin-1 gene (PS1) and promote the cleavage of amyloid precursor protein (APP) by β-secretase, resulting in the formation of amyloid plaques [22,23]. A study using transgenic mouse models confirms that hyperhomocysteinemia induces the phosphorylation of GSK-3β, significantly decreasing its activity while increasing kinase activity, thereby generating elevated Aβ levels via the γ-secretase pathway. Furthermore, dysregulation of the activities of cyclin-dependent kinase 5 (CDK5), GSK-3β, and protein phosphatase-2A (PP2A) contribute significantly to the development of tau pathology [22].
Oxidative stress: Oxidative stress serves as a potential factor in the progression from normal ageing to AD by disrupting membrane permeability, altering the cytoskeleton, causing mitochondrial damage, and leading to cell death [24,25]. Increased protein oxidation, LPO, free carbonyls, alcohol, aldehydes, and oxidative modifications in nuclear and mitochondrial DNA are well known key elements of oxidative stress [26], which induce cytotoxicity by generating free radicals and destabilising biological molecules. Additionally, oxidising agents and LPO products, such as 4-HNE, increase the activity and expression of APP, β-site amyloid precursor protein cleaving enzyme 1 (BACE1), and γ-secretase, followed by upregulation of PS-1, ultimately leading to Aβ overproduction [27].
Endoplasmic stress (ER stress): ER stress plays a critical role in various biological functions, including protein biosynthesis, maturation, proper folding, post-translational modification, and the assembly of newly synthesised proteins. The aggregation of amyloid plaques introduces deleterious effects on the ER, which can compromise its integrity and function, leading to the accumulation of unfolded proteins in its lumen. This accumulation further results in irreversible ER stress and activates the Unfolded Protein Response (UPRER) [28,29]. Additionally, the aggregation of misfolded proteins disrupts intracellular Ca2+ balance, initiating a decline in protein synthesis. During ER stress, three stress-responsive transmembrane proteins-Pancreatic ER Kinase (PERK), Activating Transcription Factor-6 (ATF-6), and Inositol-Requiring Enzyme-1 (IRE-1)-are activated to initiate the UPRER. Glucose-Regulated Protein (GRP78) also serves as a key sensor in this response. PERK facilitates the breakdown of ATF-6 (P50) and phosphorylates eukaryotic Initiation Factor 2 (eIF2α), which induces the expression of ER chaperones such as GRP78. Since ER stress is a sensitive factor that can be modulated by the overexpression of chaperone molecules, a decreased expression of these chaperones may alternatively promote the formation of Aβ-42. Moreover, overexpression of BACE-1 and increased levels of APP are significantly linked with ER stress, which in turn leads to the formation of Aβ in the temporal lobes of the cerebrum. Aβ-induced GRP78 dysregulates Ca2+ homeostasis via ER stress. This Ca2+ imbalance may promote mutations in presenilin-1/2 and activate reactive oxygen species (ROS), leading to oxidative stress and mitochondrial dysfunction, ultimately inducing cytotoxic effects [30,31].
The cholinergic hypothesis: The cholinergic hypothesis states that the neurotoxic effects of Aβ directly correlate with reduced acetylcholine (ACh) levels at the neuronal level or a defect in the biosynthesis of ACh at the synapse [32,33]. However, the concentration of ACh can be affected by a deficiency of choline acetyltransferase (ChAT), which is regarded as the key enzyme for ACh synthesis, catalysing the action of acetylcholinesterase. Acetylcholinesterase is another essential enzyme for cholinergic transmission at the synapse, as it is involved in accelerating the hydrolysis of ACh. Severe deficiencies or defects in cholinergic transmission from the basal frontal lobe can lead to presynaptic cholinergic impairment, which correlates with the severity of dementia and deterioration of memory in AD. Additionally, the interaction of presenilin-1 (PS-1) and acetylcholinesterase enhances APP expression and promotes the formation of Aβ [33].
Metal ions hypothesis: Dyshomeostasis or increased concentrations of metal ions (Copper, Zinc, Iron, Calcium) promote the formation of Aβ in AD. Furthermore, the interaction of Aβ with these metal ions introduces oxidative stress, leading to an increased production of ROS as well as disturbances in hydrogen peroxide (H2O2) levels [34]. Additionally, higher concentrations of metal ions can directly or indirectly disrupt organelles, ER stress, mitochondrial damage, autophagic dysfunction, and impairing Blood-Brain Barrier (BBB) function, including synaptic functions. Consequently, this promotes the generation of amyloid plaque formation and tau hyperphosphorylation by activating protein kinases such as secretases, GSK-3β, CDK5, and Mitogen-Activated Protein Kinases (MAPKs), while inhibiting PP2A [34].
Cholesterol transport: The regulation of lipids, including cholesterol, is a driving factor for normal brain function, synaptogenesis, and neuronal differentiation. Disruptions in phospholipids, cholesterol, or fluctuations in cholesterol concentration negatively impact the formation of amyloid peptides, BBB integrity, mitochondrial function, oxidative stress, inflammation, and neurodegeneration [35]. Moreover, ApoE4 plays a significant role in transporting cholesterol from astrocytes to neurons via lipoproteins, thereby influencing AD pathology [36]. Additionally, APOE4 affects lipid transportation between cells and Aβ clearance, which eventually promotes Aβ aggregation into toxic conformations [37]. Similarly, the oxidised form of cholesterol (oxysterol) also leads to neuroinflammation and cytotoxic effects in the brain [38]. Furthermore, free radicals generated from 7-oxocholesterols increase the production of ROS, leading to mitochondrial dysfunction, including changes in oxidative phosphorylation [39].
Association of AD and synaptic failure: Synaptic failure, a characteristic manifestation of AD, is strongly linked with Aβ deposition and tau hyperphosphorylation [40]. Moreover, extracellular glutamate accumulation and a disturbed glutamate cycle due to the toxicity of Aβ result in synaptic disturbances. Conversely, the formation of Aβ plaques also leads to glutamate spillover through α7 nicotinic ACh receptors (α7nAChR), which stimulates extrasynaptic NMDARs, ultimately contributing to neurotoxicity [40]. Additionally, previous studies have reported that Aβ oligomers activate astrocytes and increase the release of glutamate through nAChR, further stimulating extrasynaptic NMDARs [41]. Similarly, Aβ promotes the induction of extracellular glutamate, considered a detrimental factor, leading to the enhancement of extrasynaptic NMDARs, Long-Term Potentiation (LTP), Long-Term Depression (LTD), and synapse loss. Furthermore, the neurotoxicity of Aβ is also associated with the endocytosis of NMDARs in cortical neurons and a depression of NMDA-evoked currents, which, in turn, activates nAChRs and causes NMDAR agonist-induced delayed cognitive dysfunction. Additionally, the disruption between NMDAR activation and Aβ production leads to the upregulation of NMDAR overactivation and Aβ deposition, ultimately resulting in impaired synaptic plasticity and cognitive function in AD [42].
Neuroinflammation: Neuroinflammation is recognised as a significant event that induces the formation of amyloid plaques and NFTs in AD. Microglia and astrocytes play key roles in processing inflammation in the central nervous system and contribute to the pathogenesis of dementia and cognitive impairment. Furthermore, accumulated neurotoxic proteins like Aβ and tau proteins result in the release of a myriad of proinflammatory cytokines, including Tumour Necrosis Factor alpha (TNF-α) and Interleukin (IL-6). Other important protein kinases that upregulate amyloid plaques include GSK-3β, MAPK, and Cell Division Cycle 2 Kinase (CDC2K), all of which are influenced by inflammatory markers in the progression of AD and cognitive impairment [43]. Moreover, the neurotoxic effects of Aβ contribute to the release of Nitric Oxide (NO), ROS, proinflammatory cytokines, and chemokines, and vice versa. Additionally, Aβ toxicity influences astrocytes and microglia, leading to a process called reactive gliosis, which promotes the expression of an astrocyte-specific intermediate filament protein (GFAP) and allograft inflammatory factor-1 (AIF-1), a microglial-specific protein that is significantly increased in AD. Furthermore, microglial cells produce an abundance of proinflammatory cytokines, including IL-1, IL-6, and other toxic products such as ROS, NO, and cytokines, which likely enhance APP production and Aβ formation. In addition, the deposition of Aβ may interact with various microglial receptors like CD14, CD36, and CD47, while inflammatory activity occurs through the binding of Aβ to CD36 [44].
Discussion
The Aβ peptide can interact with potential receptors and activate downstream pathways, leading to the generation of oxidative stress and ROS. This, in turn, induces the hyperphosphorylation of Tau protein, activates microglial and astrocyte cells, and triggers inflammatory responses, which may result in neuronal death and contribute to AD. The neurotoxic effects of Aβ and its possible interlinking with other biological markers have been compared in [Table/Fig-3] [45-52].
Illustrates the comparative view of neurotoxic effects of Amyloid Beta (Aβ) and it’s possible interlinking with other biological markers [45-52].
| Author(s), Publication year | Study place | Sample size | Outcome of study |
|---|
| Torres LL et al., [45], 2011 | Romania | 45 | A study evaluated the enzymatic markers of oxidative stress like Superoxide Dismutase (SOD) and Glutathione Peroxidase (GPX), as well as Lipid Peroxidation (LPO) markers like Malondialdehyde (MDA) in serum level were decreased for Mild Cognitive Impairment (MCI) and AD patients, compared with age-matched healthy controls. Further, they concluded enzymatic markers of oxidative damage could be key components to develop the AD pathology. |
| Padurariu M et al., [46], 2010 | Brazil | 29 | A study found that increased peripheral oxidative stress markers and reduced antioxidant enzymatic activities were positively linked with memory impairment and Aβ in AD. Additionally, an increase in plasma MDA levels and a decreased glutathione reductase/GPX ratio were also noticed in MCI and AD patients. |
| Zhang H et al., [47], 2012 | USA | 30 | A study in transgenic mice model advocated formation of protein mixed disulphide (Pr-SSG) might be an indicator for the formation of amyloid plaque. Increased mixed-disulfides in the hippocampus might be the possible inducing element to cause increasing oxidative damage under oxidising conditions [48]. |
| Vishnu VY et al., [48], 2017 | India | 68 | Previous study witnessed that proinflammatory markers (IL-6 and CRP) were not significantly associated with Aβ formation in AD, but the plasma fibrinogen was remarkably linked with vascular dementia. |
| Rani P et al., [49], 2017 | India | 45 | The study revealed formation of Aβ in AD is directly correlated with LPO. Further, Aβ also evolves into a peroxidant which induces increased oxidative stress and LPO. Additionally, Aβ also strongly linked with the Hydroxy-Non-Enal (HNE) MDA and other pathological markers in AD. |
| Atluri VSR et al., [50], 2019 | USA | 84 | Recent study revealed deposition of Aβ in the brain is one of the key hallmarks of AD pathology which might be further influenced by neuroinflammatory processes such as Nucleotide-binding oligomerisation domain, leucine-rich repeat and pyrin domain containing 3 (NLRP3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFm-κB) considered as the principle neuroinflammatory pathways to accelerate the AD pathogenesis. |
| Wilczynska K et al., [51], 2021 | Ploand | 60 | A study found significant association between inflammatory biomarker (YKL-40) and t-tau Aβ1-42/Aβ1-40) and with the severity of cognitive decline. |
| Foley KE et al., [52], 2024 | USA | 837 | A recent finding found significant association between the plasma biomarkers (Aβ40, Aβ42, Aβ42/40, ptau181, total tau, and NFL) and the peripheral inflammatory biomarkers such as (TNFα, IL6, IL8, IL10, and GFAP) in the Late Onset of Alzheimer’s Disease (LOAD). |
Conclusion(s)
The Aβ peptide is considered a key biological marker for understanding the pathomechanism of AD. Since AD is a complex phenomenon caused by multiple factors, several biochemical markers, such as the hyperphosphorylation of tau protein and oxidative damage, have been established to varying extents to accelerate the pathogenesis of AD. Determining key biological markers, such as Aβ, is essential for understanding the pathomechanism of AD and is crucial for developing novel therapeutics and future management strategies for the disease. Aβ, as an ideal biomarker, must be identified in disease candidates at the earliest stages of the disease through minimally invasive or non invasive approaches, which could be beneficial in slowing down the progression of AD.
[1]. The American Psychiatric Publishing Textbook of Alzheimer Disease and Other Dementias 2009 American Psychiatric Pub:488-491. [Google Scholar]
[2]. GBD 2019 Dementia Forecasting CollaboratorsEstimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019Lancet Public Health 2022 7(2):e105-25. [Google Scholar]
[3]. Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkersLancet Neurol 2013 12(2):207-16. [Google Scholar]
[4]. Hebert LE, Weuve J, Scherr PA, Evans DA, Alzheimer disease in the United States (2010-2050) estimated using the 2010 censusNeurology 2013 80(19):1778-83. [Google Scholar]
[5]. Wolters FJ, van der Lee SJ, Koudstaal PJ, van Duijn CM, Hofman A, Ikram MK, Parental family history of dementia in relation to subclinical brain disease and dementia riskNeurology 2017 88(17):1642-49. [Google Scholar]
[6]. Qiu C, Kivipelto M, von Strauss E, Epidemiology of Alzheimer’s disease: Occurrence, determinants, and strategies toward interventionDialogues Clin Neurosci 2009 11(2):111-28. [Google Scholar]
[7]. Livingston G, Huntley J, Sommerlad A, Ames D, Ballard C, Banerjee S, Dementia prevention, intervention, and care: 2020 report of the Lancet CommissionLancet Lond Engl 2020 396(10248):413-46. [Google Scholar]
[8]. Alzheimer’s Association ReportAlzheimer’s disease facts and figuresAlzheimer’s Dement 2023 19(4):1598-695. [Google Scholar]
[9]. Hardy JA, Higgins GA, Alzheimer’s disease: The amyloid cascade hypothesisScience 1992 256(5054):184-85. [Google Scholar]
[10]. Huang YR, Liu RT, The toxicity and polymorphism of β-amyloid oligomersInt J Mol Sci 2020 21(12):4477 [Google Scholar]
[11]. Penke B, Szűcs M, Bogár F, Oligomerization and conformational change turn monomeric β-amyloid and tau proteins toxic: Their role in Alzheimer’s pathogenesisMol Basel Switz 2020 25(7):1659 [Google Scholar]
[12]. Flagmeier P, De S, Michaels TCT, Yang X, Dear AJ, Emanuelsson C, Direct measurement of lipid membrane disruption connects kinetics and toxicity of Aβ42 aggregationNat Struct Mol Biol 2020 27(10):886-91. [Google Scholar]
[13]. Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Amyloid-β oligomerization in Alzheimer dementia versus high-pathology controlsAnn Neurol 2013 73(1):104-19. [Google Scholar]
[14]. Ji XR, Cheng KC, Chen YR, Lin TY, Cheung CHA, Wu CL, Dysfunction of different cellular degradation pathways contributes to specific β-amyloid42-induced pathologiesFASEB J Off Publ Fed Am Soc Exp Biol 2018 32(3):1375-87. [Google Scholar]
[15]. Tolar M, Hey J, Power A, Abushakra S, Neurotoxic soluble amyloid oligomers drive alzheimer’s pathogenesis and represent a clinically validated target for slowing disease progressionInt J Mol Sci 2021 22(12):6355 [Google Scholar]
[16]. Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptorNature 1987 325(6106):733-36. [Google Scholar]
[17]. Tanzi RE, Gusella JF, Watkins PC, Bruns GA, St George-Hyslop P, Van Keuren ML, Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locusScience 1987 235(4791):880-84. [Google Scholar]
[18]. Aksenov MYu, Aksenova MV, Harris ME, Hensley K, Butterfield DA, Carney JM, Enhancement of beta-amyloid peptide A beta(1-40)-mediated neurotoxicity by glutamine synthetaseJ Neurochem 1995 65(4):1899-902. [Google Scholar]
[19]. Yatin SM, Yatin M, Aulick T, Ain KB, Butterfield DA, Alzheimer’s amyloid beta-peptide associated free radicals increase rat embryonic neuronal polyamine uptake and ornithine decarboxylase activity: Protective effect of vitamin ENeurosci Lett 1999 263(1):17-20. [Google Scholar]
[20]. Mark RJ, Hensley K, Butterfield DA, Mattson MP, Amyloid beta-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell deathJ Neurosci Off J Soc Neurosci 1995 15(9):6239-49. [Google Scholar]
[21]. Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activityNature 2001 414(6860):212-16. [Google Scholar]
[22]. Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Homocysteine potentiates beta-amyloid neurotoxicity: Role of oxidative stressJ Neurochem 2001 78(2):249-53. [Google Scholar]
[23]. Zhuo JM, Praticò D, Acceleration of brain amyloidosis in an Alzheimer’s disease mouse model by a folate, vitamin B6 and B12-deficient dietExp Gerontol 2010 45(3):195-201. [Google Scholar]
[24]. Perry G, Nunomura A, Hirai K, Takeda A, Aliev G, Smith MA, Oxidative damage in Alzheimer’s disease: The metabolic dimensionInt J Dev Neurosci Off J Int Soc Dev Neurosci 2000 18(4-5):417-21. [Google Scholar]
[25]. Hernández-Rodríguez M, Arciniega-Martínez IM, García-Marín ID, Correa-Basurto J, Rosales-Hernández MC, Chronic administration of scopolamine increased GSK3βP9, beta secretase, amyloid beta, and oxidative stress in the hippocampus of wistar ratsMol Neurobiol 2020 57(9):3979-88. [Google Scholar]
[26]. Zhang JS, Zhou SF, Wang Q, Guo JN, Liang HM, Deng JB, Gastrodin suppresses BACE1 expression under oxidative stress condition via inhibition of the PKR/eIF2α pathway in Alzheimer’s diseaseNeuroscience 2016 325:01-09. [Google Scholar]
[27]. Sun X, Bromley-Brits K, Song W, Regulation of β-site APP-cleaving enzyme 1 gene expression and its role in Alzheimer’s diseaseJ Neurochem 2012 120(Suppl 1):62-70. [Google Scholar]
[28]. Goswami P, Afjal MA, Akhter J, Mangla A, Khan J, Parvez S, Involvement of endoplasmic reticulum stress in amyloid β (1-42)-induced Alzheimer’s like neuropathological process in rat brainBrain Res Bull 2020 165:108-17. [Google Scholar]
[29]. Ghemrawi R, Khair M, Endoplasmic reticulum stress and unfolded protein response in neurodegenerative diseasesInt J Mol Sci 2020 21(17):6127 [Google Scholar]
[30]. Ohno M, PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s diseaseBrain Res Bull 2018 141:72-78. [Google Scholar]
[31]. Duran-Aniotz C, Cornejo VH, Espinoza S, Ardiles ÁO, Medinas DB, Salazar C, IRE1 signaling exacerbates Alzheimer’s disease pathogenesisActa Neuropathol (Berl) 2017 134(3):489-506. [Google Scholar]
[32]. Bekdash RA, The cholinergic system, the adrenergic system and the neuropathology of Alzheimer’s diseaseInt J Mol Sci 2021 22(3):1273 [Google Scholar]
[33]. Salim S, Oxidative stress and the central nervous systemJ Pharmacol Exp Ther 2017 360(1):201-05. [Google Scholar]
[34]. Wang L, Yin YL, Liu XZ, Shen P, Zheng YG, Lan XR, Current understanding of metal ions in the pathogenesis of Alzheimer’s diseaseTransl Neurodegener 2020 9(1):10 [Google Scholar]
[35]. Chew H, Solomon VA, Fonteh AN, Involvement of lipids in Alzheimer’s disease pathology and potential therapiesFront Physiol 2020 11:598 [Google Scholar]
[36]. Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G, Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapyNat Rev Neurol 2013 9(2):106-18. [Google Scholar]
[37]. Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell typesNeuron 2018 98(6):1294 [Google Scholar]
[38]. Loera-Valencia R, Vazquez-Juarez E, Muñoz A, Gerenu G, Gómez-Galán M, Lindskog M, High levels of 27-hydroxycholesterol results in synaptic plasticity alterations in the hippocampusSci Rep 2021 11(1):3736 [Google Scholar]
[39]. Lanfranco MF, Ng CA, Rebeck GW, ApoE lipidation as a therapeutic target in Alzheimer’s diseaseInt J Mol Sci 2020 21(17):6336 [Google Scholar]
[40]. Huang S, Tong H, Lei M, Zhou M, Guo W, Li G, Astrocytic glutamatergic transporters are involved in Aβ-induced synaptic dysfunctionBrain Res 2018 1678:129-37. [Google Scholar]
[41]. Talantova M, Sanz-Blasco S, Zhang X, Xia P, Akhtar MW, Okamoto S, Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic lossProc Natl Acad Sci U S A 2013 110(27):E2518-E2527. [Google Scholar]
[42]. Zhang H, Jiang X, Ma L, Wei W, Li Z, Chang S, Role of Aβ in Alzheimer’s-related synaptic dysfunctionFront Cell Dev Biol 2022 10:964075 [Google Scholar]
[43]. Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Caspase signalling controls microglia activation and neurotoxicityNature 2011 472(7343):319-24. [Google Scholar]
[44]. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammationNat Immunol 2013 14(8):812-20. [Google Scholar]
[45]. Torres LL, Quaglio NB, de Souza GT, Garcia RT, Dati LMM, Moreira WL, Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s diseaseJ Alzheimers Dis JAD 2011 26(1):59-68. [Google Scholar]
[46]. Padurariu M, Ciobica A, Hritcu L, Stoica B, Bild W, Stefanescu C, Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s diseaseNeurosci Lett 2010 469(1):06-10. [Google Scholar]
[47]. Zhang C, Rodriguez C, Spaulding J, Aw TY, Feng J, Age-dependent and tissue-related glutathione redox status in a mouse model of Alzheimer’s diseaseJ Alzheimers Dis JAD 2012 28(3):655-66. [Google Scholar]
[48]. Vishnu VY, Modi M, Garg VK, Mohanty M, Goyal MK, Lal V, Role of inflammatory and hemostatic biomarkers in Alzheimer’s and vascular dementia-A pilot study from a tertiary center in Northern IndiaAsian J Psychiatry 2017 29:59-62. [Google Scholar]
[49]. Rani P, Krishnan S, Rani Cathrine C, Study on analysis of peripheral biomarkers for Alzheimer’s disease diagnosisFront Neurol 2017 8:328 [Google Scholar]
[50]. Atluri VSR, Tiwari S, Rodriguez M, Kaushik A, Yndart A, Kolishetti N, Inhibition of Amyloid-Beta production, associated neuroinflammation, and histone deacetylase 2-mediated epigenetic modifications prevent neuropathology in Alzheimer’s disease in vitro modelFront Aging Neurosci 2019 11:342 [Google Scholar]
[51]. Wilczyńska K, Maciejczyk M, Zalewska A, Waszkiewicz N, Serum amyloid biomarkers, tau protein and YKL-40 utility in detection, differential diagnosing, and monitoring of dementiaFront Psychiatry 2021 12:725511 [Google Scholar]
[52]. Foley KE, Winder Z, Sudduth TL, Martin BJ, Nelson PT, Jicha GA, Alzheimer’s disease and inflammatory biomarkers positively correlate in plasma in the UK-ADRC cohortAlzheimers Dement J Alzheimers Assoc 2024 20(2):1374-86. [Google Scholar]