Emanuel Syndrome (ES) is a chromosomal disorder characterised by the presence of an extra copy of chromosome 22, specifically a derivative 22 chromosome, which results from an unbalanced translocation involving chromosomes 11 and 22, due to 3:1 meiotic non disjunction. This leads to the gain of the 11q23-qter and 22pter-q11.2 regions. The syndrome is marked by developmental delay, facial dysmorphism, heart defects, genital abnormalities and renal anomalies. In most cases, one of the parents is a carrier of a balanced translocation, t(11;22). The present case is of a two-month-old male child suffering from failure to thrive and developmental delay, who was found to have a karyotype of 47,XY,+der(22)t(11;22)(q23.3;q11.2)dmat, resulting from a maternal balanced translocation, t(11;22)(q23;q11.2). Molecular cytogenetic testing confirmed the presence of partial trisomy 22q11.2 in the proband. This report presents a case of partial trisomy 22q11.2 resulting from a maternal balanced translocation between the long arms of chromosomes 11 and 22, associated with Congenital Heart Defects (CHD) and Congenital Diaphragmatic Hernia (CDH). Approximately, 11 comparable cases have been reported in the Indian population, with variations in breakpoints observed in some. However, none of these previous cases have identified CDH as a phenotypic feature, making this case particularly noteworthy.
Case Report
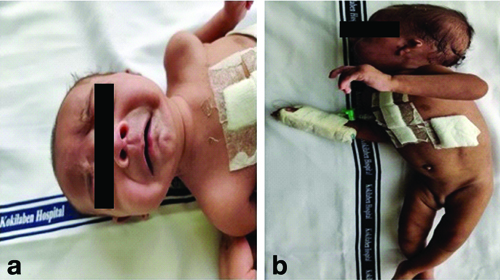
A male child was delivered by Lower Segment Caesarean Section (LSCS) due to prolonged labour at 38 weeks of gestation, weighing 2.3 kg. The parents, who are non consanguineous (the father is 42 years old and the mother is 33 years old), have a history of one first-trimester miscarriage. The case was referred to our hospital when the child was two months and ten days old due to chief complaints of excessive crying, irritability, and failure to thrive since one month of age. The child exhibited syndromic features like significant hypotonia, cleft palate, developmental delay, difficulty in breathing and feeding, left microtia, micrognathia, retrognathia, grossly abnormal bone structure, and undescended testis [Table/Fig-1].
a) Clinical features of the proband indicating microcephaly, micrognathia and retrognathia; b) Clinical features of the proband indicating left microtia, scissoring of lower limbs and undescended testes.
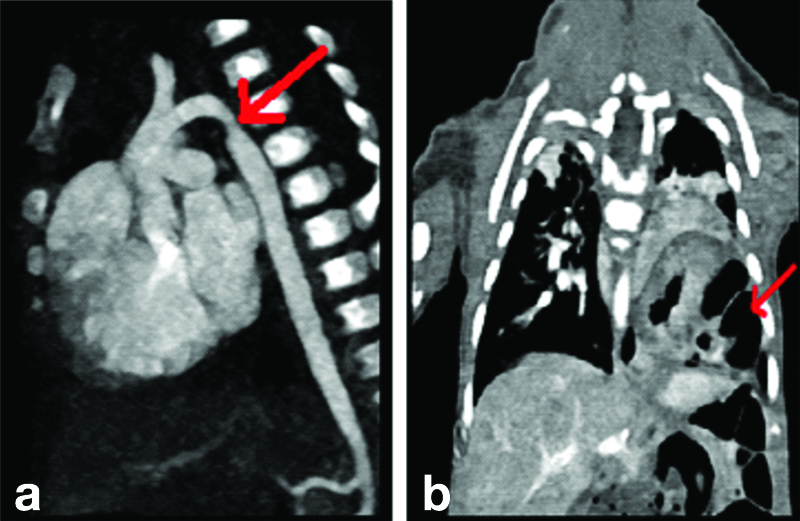
The 2D Echo showed suspected coarctation of the aorta. The child also exhibited tachypnea and tachycardia. Upon further examination, he was found to have CHD with an Ostium Secundum (OS) and Atrial Septal Defect (ASD). A Computed Tomography (CT) angiogram suggested a mildly hypoplastic aortic arch and a left-sided diaphragmatic hernia [Table/Fig-2]. The child underwent surgery for these conditions. However, two months later, he experienced respiratory failure and was placed on mechanical ventilation. Gradually, the ventilator settings were increased, and he was switched to High-Frequency Oscillatory Ventilation (HFOV).
Image of CT with angiography indicating: (a) Mild hypoblastic arch; (b) Left-sided diaphragmatic hernia.
First-line management for Pulmonary Arterial Hypertension (PAH) was initiated. Nevertheless, the child’s condition continued to deteriorate. He developed Acute Kidney Injury (AKI), which was managed with optimal fluid balance and diuretics. Repeat 2D Echo were conducted alongside ASD repair, and he was subsequently transferred to the cardiac Intensive Care Unit (ICU) on HFOV. During the ASD repair, it was discovered that the CDH had recurred, and the child was simultaneously operated for the same. Postoperatively, he continued on HFOV.
After five days, the child was shifted to conventional ventilation, and two days later, he was extubated to Continuous Positive Airway Pressure (CPAP). The child tolerated extubation well and was gradually weaned off CPAP. A postoperative 2D Echo showed mild PAH. With chest physiotherapy and optimisation of antibiotics, the child was eventually sent to the ward. Currently, at the age of two years, the child is doing well developmentally.
Cytogenetic Findings
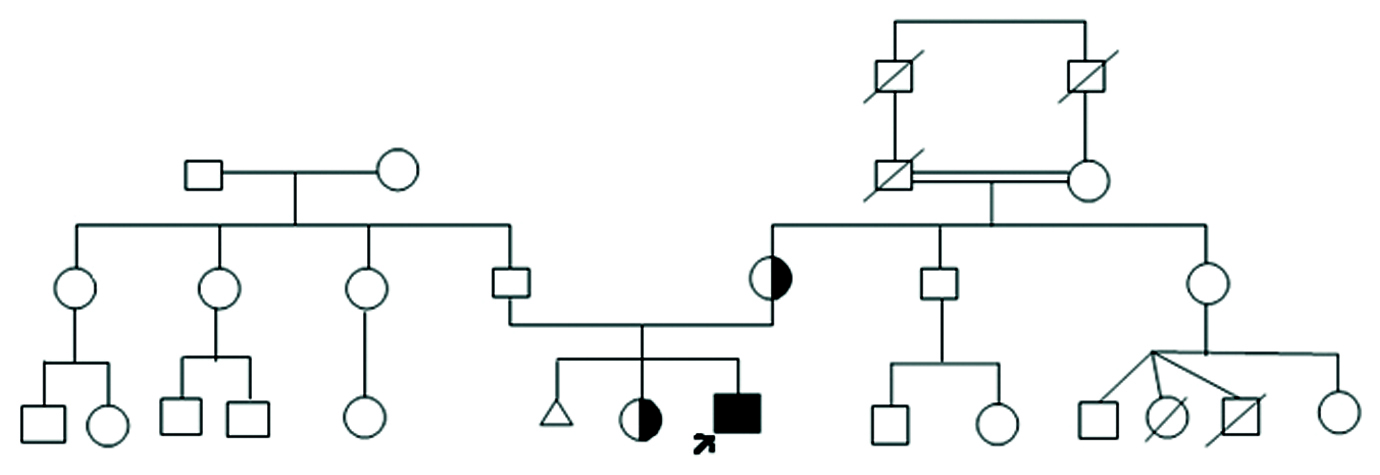
In view of CHD and CDH, a peripheral blood sample was sent to the Department of Genetics and Molecular Medicine for Fluorescence In Situ Hybridisation (FISH) evaluation to rule out DiGeorge syndrome, specifically the deletion of 22q11.2. The FISH results were negative for DiGeorge syndrome; instead, a gain of the 22q11.2 region was observed. A detailed pedigree chart was created [Table/Fig-3].
The family pedigree of the proband.
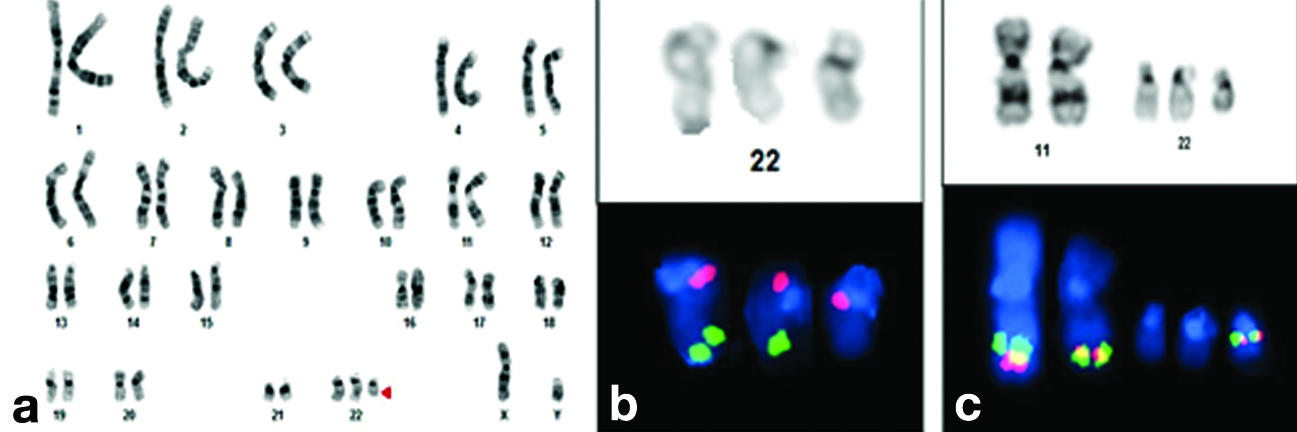
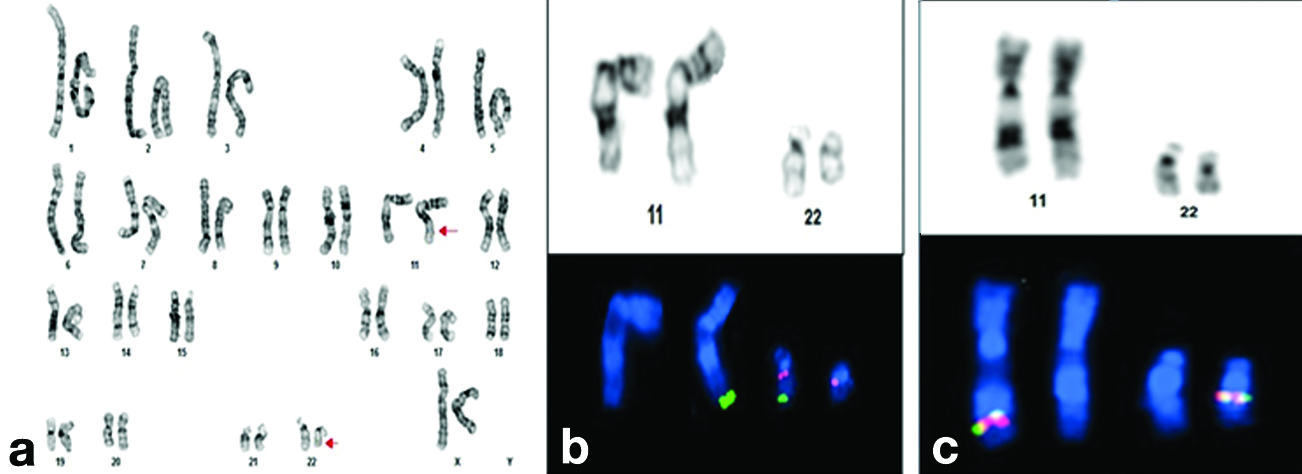
Conventional cytogenetic analysis was performed using PHA-stimulated 72-hour lymphocyte cultures, followed by GTG banding techniques at 550-band resolution. This analysis revealed an extra derivative chromosome 22, resulting in a karyotype of 47,XY,+der(22), t(11;22)(q23.3;q11.2) [Table/Fig-4]. Parental karyotyping was conducted to determine the origin of the extra derivative chromosome 22. The maternal karyotype was found to have a reciprocal balanced translocation involving chromosomes 11 and 22, with the karyotype being 46,XX,t(11;22)(q23.3;q11.2) [Table/Fig-5]. The father’s karyotype was normal. Karyotype analysis of the elder sister of the proband also indicated a reciprocal balanced translocation involving chromosomes 11 and 22, similar to that of the mother.
a) GTG banded karyotype of proband indicating partial trisomy of chromosome 22; b) Partial karyotype image of sequential FISH on G-banded metaphase of proband using Vysis LSI TUPLE (HIRA)/LSI ARSA probe indicating an extra red signal on supernumerary chromosome. Green: 22q13.3(ARSA); Orange: 22q11.2(HIRA); c) Partial karyotype image of sequential FISH on G-banded metaphase of proband using Vysis LSI MLL dual colour break apart rearrangement probe (11q23 MLL SpectrumGreen-SpectrumOrange).
a) GTG banded karyotype of proband’s mother showing balanced translocation between chromosome 11 and chromosome 22; b) Partial karyotype image of sequential FISH on G-banded metaphase of proband’s mother using Vysis LSI TUPLE(HIRA)/LSI ARSA probe. Green: 22q13.3(ARSA);Orange:22q11.2(HIRA); c) Partial karyotype image of sequential FISH on G-banded metaphase of proband using Vysis LSI MLL dual colour break apart rearrangement probe (11q23 MLL SpectrumGreen-SpectrumOrange).
The parents were counselled and explained about the recurrence risk of the disorder. Prenatal diagnosis for the elder sister of the proband in her future pregnancies was recommended as a preventive measure.
Discussion
The ES, also known as derivative 22 syndrome, is an unbalanced translocation resulting from the 3:1 malsegregation of the parental derivative 22 during gametogenesis. Patients with ES present with distinctive phenotypes, including severe intellectual disability, microcephaly, preauricular tags or pits, failure to thrive, cleft or high-arched palates, micrognathia, kidney abnormalities, CHD, and genital abnormalities in males [1]. Typically, one parent carries a balanced reciprocal translocation, t(11;22), and is phenotypically normal. After experiencing several miscarriages, infertility issues, or the birth of a child with supernumerary derivative 22 syndrome, carrier status is often determined [2]. The unbalanced offspring exhibit a duplication of chromosomes 22 and 11q. Some patients with this chromosomal abnormality are classified as having partial trisomy 22, while others are referred to as having partial trisomy 11q. The alternative product of the 3:1 meiotic disjunction {45,XY,-22,-11,+der(11)} has been documented only once. In all these families, the translocation t(11;22)(q23;q11) appears to be identical [1].
It has been observed that female carriers of t(11;22) have an estimated 4% risk of having children with the supernumerary der(22), while male carriers have a lower risk of 0.7% [3]. The prevalence of the rare ES remains unknown, but based on the frequency of de novo t(11;22) translocations in sperm from healthy men, the estimated occurrence in the general population is 1 in 110,000. The highest mortality rates are observed in the first few months of life [3,4]. For carriers identified after the birth of a child with chromosomal imbalance, the risk of recurrence in subsequent pregnancies ranges from 5 to 10% [3]. Balanced chromosomal translocation carriers may produce gametes with unbalanced chromosomal translocations during gametogenesis, leading to recurrent spontaneous abortions or congenital abnormalities in their offspring [5]. Carriers of the t(11;22) translocation usually show no symptoms because it does not affect functional genes. However, they often face reproductive issues like male infertility and recurrent pregnancy loss, and they are at a higher risk for chromosomal imbalances in their offspring [4]. Severely affected offspring develop supernumerary der(22)t(11;22) syndrome (ES) due to a 3:1 meiotic malsegregation. The incidence of 3:1 segregation is notably higher in embryos from female translocation carriers [6].
The chromosome 22q region is prone to rearrangements that cause congenital disorders such as cat-eye syndrome, der(22) syndrome, and VCFS/DGS due to tetrasomy, trisomy, or monosomy. The breakpoints occur within a low copy repeat known as the "LCR22" region. Edelmann L et al., described how LCR22s midiate homologous recombination events, leading to various rearrangements associated with these disorders. In the t(11;22) translocation, the breakpoint on 22q11 falls within one of the nine LCR22s that cover the 22q11 region [7]. Edelmann L et al., explained the phenomenon of AT-rich repeats within the region of 11q23, which constitutes a minisatellite or Variable Number of Tandem Repeats (VNTR), an unstable region of the genome that leads to chromosomal rearrangements, including reciprocal translocation. LCR22s include AT rich repeats that could contribute to the rearrangements observed on chromosomes 11 and 22. They also proposed that the AT-rich repeats in LCR22s facilitate recombination events between non homologous chromosomes during meiosis, leading to 22q11 deletions and duplications associated with congenital anomaly disorders. However, the exact mechanism remains unknown [8].
Although all the cases reported in the Indian population exhibited a similar abnormality, variations in the breakpoint and clinical features were observed in some cases [Table/Fig-6] [3,9-13]. Kamath V et al., reported three cases of +der(22)t(11;22)(q23-23.3;q11.2) with no history of consanguineous marriage among their parents. These cases exhibited dysmorphism, developmental delay, and various congenital abnormalities, including two atrial and one ventricular cardiac septal defect, hearing impairment, seizures (two occurrences each), hypotonia, visual loss, and genital abnormalities [3]. Tiriya D et al., reported a case with features of high arched palate, micrognathia, preauricular skin tags, microcephaly, a sacral dimple, hypotonia, and OS ASD [9]. Similarly, Choudhary M et al., and Shenoy RD et al., also reported cases of +der(22)t(11;22) [10,11]. Comparatively, Saxena D et al., reported one case with a 47,+mar karyotype and a gain of 18 Mb on chromosome 11, along with a gain of 3.4 Mb on chromosome 22 as identified through microarray, in which almost all clinical features were absent except for facial dysmorphism [12]. However, Deepika MLN et al., reported a case of a single foetus at 17-18 weeks gestation with intrauterine growth restriction, dysplastic ears, and CHD including a small Ventricular Septal Defect (VSD) and moderate Patent Ductus Arteriosus (PDA), confirmed by 2D Echo and sonography suggestive of ES. However, no genetic confirmation was performed [13]. There was a slight variance in the breakpoints of the cases reported by Shenoy RD et al., and Saxena D et al., specifically at 11q24-q25 and 22q12-q13.1, compared to the other cases and present case, but microarray analysis showed approximately similar Mb gains (18 Mb-18.2 Mb) on chromosome 11 [11,12]. In all the cases, the abnormality was of maternal origin, except in one case where parental karyotyping was not performed [13]. Most of the cases presented with CHD, but none exhibited CDH as a clinical feature [Table/Fig-6].
List of reported cases of Emanuel Syndrome (ES) from India.
| Clinical features | Kamath V et al., [3] | Tiriya D et al., [9] | Choudhary M et al., [10] | Saxena RD et al., [12] | Shenoy RD et al., [11] | Deepika MLN et al., [13] | Present case |
|---|
| Developmental delay | + | + | + | - | + | - | + | + | - | - | - | + |
| Facial dysmorphism | + | - | - | - | + | - | - | - | - | - | + | - |
| Frontal bossing | - | - | - | - | + | + | + | - | - | - | - | - |
| Sparse hair | - | - | - | - | - | + | - | - | - | - | - | - |
| Sparse eyebrows | - | - | - | - | - | + | + | + | + | - | - | - |
| Unilateral ptosis | - | - | - | - | - | - | - | - | - | + | - | - |
| Downward slant eyes | - | - | - | - | - | + | + | + | + | + | - | - |
| Preauricular pit | - | - | - | + | - | + | - | + | + | - | - | - |
| Long philtrum | - | - | - | - | + | + | - | + | - | - | - | - |
| Downturned mouth corners | - | - | - | - | + | + | - | - | - | - | - | - |
| Micrognathia | - | - | - | + | + | + | + | - | - | + | | + |
| Ear anomalies | - | - | - | - | + | | | + | - | + | + | + |
| Hearing impairment | + | + | - | - | + | - | - | - | - | - | - | - |
| Visual loss | + | - | - | - | - | - | - | - | - | - | - | - |
| Severe intellectual disability | - | - | - | - | - | - | - | - | - | - | - | - |
| Microcephaly | - | - | + | + | - | - | - | - | - | + | - | + |
| Failure to thrive | | - | - | - | - | - | - | - | - | - | - | + |
| Cleft palate | + | - | - | - | - | - | - | - | + | - | - | + |
| High-arched palate | | - | - | + | + | - | - | - | - | - | - | - |
| Hypotonia | + | - | - | + | + | - | - | - | - | + | - | - |
| Renal abnormalities | - | - | - | - | + | - | - | - | - | - | + | - |
| Congenital Heart Defects (CHD) | + | + | + | + | + | - | - | + | + | - | + | + |
| Genital abnormalities-Undescended testis | - | + | - | - | + | - | - | - | - | - | - | + |
| Intrauterine growth restriction | - | - | - | - | - | - | - | - | - | - | + | - |
| Diaphragmatic hernia | - | - | - | - | - | - | - | - | - | - | - | + |
| Carrier status | Mother | Mother | Mother | Mother | Mother | Mother | Mother | Mother | Mother | Mother | Not done | Mother |
| Consanguinity | No | No | No | No | Yes | No | No | No | No | No | Yes | No |
| Cytogenomics |
| Karyotype of proband | 47,XX,+der(22)t(11;22)(q23;q11.2)mat | 47,XY,+der(22)t(11;22)(q23;q11.2)mat | 47,XX,+der(22)t(11;22)(q23;q11.2)mat | 47,XY,+der(22), t(11;22)(q23;q11.2) | 47,XX,+der(22), t(11;22) | 47,+mar | 47,+mar | 47,+mar | 47,XX,+der(22), t(11;22) | 47,XY,+der(22), t(11;22) (q24; q12) | Not done | 47,XY,+der(22), t(11;22) (q23;q11.2) dmat |
| FISH | Done | Done | Done | Not done | Not done | 22q11.2 trisomy | 22q11.2 trisomy | Not done | Not done | Done | Not done | Done |
| Microarray | Not done | Not done | Not done | Not done | Not done | Gain of 18Mb at 11q and 3.4 Mb at 22q | Gain of 18Mb at 11q and 3.6 Mb at 22q | Not done | Not done | gain of 18.2 Mb on chromosome 11 and gain of 4.6 Mb on chromosome 22 | Not done | Not done |
| Karyotype ofcarrier parent | 46,XX,t(11;22)(q23;q11.2) | 46,XX,t(11;22)(q23;q11.2) | 46,XX,t(11;22)(q23;q11.2) | 46,XX,t(11;22)(q23;q11.2) | 46,XX,t(11;22)(q23.3;q11.2) | 46,XX,t(11;22)(q23:q11) | 46,XX,t(11;22)(q25;q13.1) | 46,XX, t(11;22)(q25;q13) | 46,XX, inv(9), t(11;22) | 46,XX, t(11;22) (q24; q12) | Not done | 46,XX, t(11;22)(q23.3;q11.2) |
Genetic counselling is crucial for families with a carrier of the t(11;22) translocation. Future pregnancies carry an increased risk for ES, the balanced t(11;22) translocation, or other meiotic malsegregations. Prenatal cytogenetic testing is recommended for these pregnancies. Unaffected siblings should also consider carrier testing in adulthood [12]. If a sibling carries the balanced t(11;22), genetic counselling can help them understand potential risks to their children and guide reproductive decisions. For individuals with ES, multidisciplinary care and regular developmental assessments are essential, tailored to their age and systemic involvement.
Conclusion(s)
Present case described that partial trisomy 22 is characterised by a recognisable pattern of CHD and CDH. This study confirms the typical phenotype associated with partial trisomy 22 and underscores its connection to congenital anomalies. Early diagnosis through cytogenetic testing is crucial for planning effective management of CHD and CDH. Advanced molecular cytogenetic techniques, such as chromosomal microarray, offer precise diagnostic information, aiding in prognosis and tailored management of affected individuals. The study also emphasises the importance of genetic counselling and detailed family pedigree charting in diagnosis, as well as in assessing the occurrence/reoccurrence of the condition and in preventing its reoccurrence.
[1]. Zackai EH, Emanuel BS, Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunctionAm J Med Genet 1980 7:507-21. [Google Scholar]
[2]. Carter MT, Barrowman NJ, St.Pierre SA, Emanuel BS, Boycott KM, Risk of breast cancer not increased in translocation 11;22 carriers: Analysis of 80 pedigreesAm J Med Genet A 2010 152A:212-14. [Google Scholar]
[3]. Kamath V, Srivastava VM, Yuvarani S, Chacko MP, Bhattacharya SK, Oommen SP, The constitutional balanced translocation t(11;22)(q23;q11.2)-An Indian accountJ Clin Diagn 2019 13:GC01-GC04. [Google Scholar]
[4]. Ohye T, Inagaki H, Kato T, Tsutsumi M, Kurahashi H, Prevalence of emanuel syndrome: Theoretical frequency and surveillance resultPediatr Int 2014 56(4):462-66. [Google Scholar]
[5]. Sanyal D, Bhairi V, Kadandale JS, Practice of consanguinity and unusual cases of inherited familial chromosome abnormalities: A case reportInt J Mol Cell Med 2016 5(1):57-63. [Google Scholar]
[6]. Lledó B, Ortiz JA, Morales R, Ten J, de la Fuente PE, García-Ochoa C, The paternal effect of chromosome translocation carriers observed from meiotic segregation in embryosHum Reprod 2010 25(7):1843-48. [Google Scholar]
[7]. Edelmann L, Pandita RK, Spiteri E, Funke B, Goldberg R, Palanisamy N, A common molecular basis for rearrangement disorders on chromosome 22q11Hum Mol Genet 1999a 8:1157-67. [Google Scholar]
[8]. Edelmann L, Spiteri E, McCain N, Goldberg R, Pandita R, Duong S, A common breakpoint on 11q23 in carriers of the constitutional t(11;22) translocationAm J Hum Genet 1999b 65(6):1608-16. [Google Scholar]
[9]. Tiriya D, Rathod D, Pawar D, Suryavanshi D, Khaire D, Emanuel syndrome: A rare case reportInd J Basic Appl Med Res 2020 9(3):355-57. [Google Scholar]
[10]. Choudhary M, Babaji P, Sharma N, Dhamankar D, Naregal G, Reddy V, Derivative 11;22 (Emanuel) syndrome: A case report and a reviewJ Pediatr Surg Case Rep 2013 9:01-04. [Google Scholar]
[11]. Shenoy RD, Shenoy V, Shetty V, Chromosomal abnormalities in syndromic orofacial clefts: Report of three childrenCase Rep Genet 2018 2018:1928918 [Google Scholar]
[12]. Saxena D, Srivastava P, Tuteja M, Mandal K, Phadke S, Phenotypic characterization of derivative 22 syndrome: Case series and reviewJ Genet 2018 97(1):205-11. [Google Scholar]
[13]. Deepika MLN, Sunitha T, Srinadh B, Prasoona KR, Sujatha M, Madireddi S, Prenatal assessment of three rare syndromes from Telangana region by 3D/4D sonographyJ Genet Syndr Gene Ther 2016 7(5):1000309 [Google Scholar]