Holt-Oram syndrome is a rare congenital autosomal dominant disorder caused by a mutation in the TBX5 gene. It is characterised by upper limb abnormalities and congenital heart lesions, such as Atrial Septal Defect (ASD), typically affecting children. In this case, a six-year-old boy presented with facial puffiness and decreased urine output following an upper respiratory infection. Upon examination, he was found to have Holt-Oram syndrome, a rare genetic disorder characterised by abnormalities in the bones of the upper limbs and congenital heart defects. He developed acute Post Streptococcal Glomerulonephritis (PSGN) following a streptococcal infection, resulting in persistent hypertension. Each condition independently poses significant health challenges, and their simultaneous occurrence complicates the clinical picture considerably. Following detailed investigations and management, the child required continued treatment with two antihypertensive medications upon discharge. The need for dual therapy in a paediatric patient highlights the severity of his condition and the challenges in managing it effectively. Close follow-up is essential to monitor disease progression and ensure optimal outcomes. This case features both a genetic disorder (Holt-Oram syndrome due to a TBX5 gene mutation) and an immune-mediated renal disease (PSGN), representing a rare intersection of genetic and postinfectious pathologies. The co-existence of these two conditions is unusual and complicates the clinical picture, necessitating a tailored approach to management. This case underscores the need for a multidisciplinary approach involving cardiology, nephrology, and genetics. Coordinated care is crucial for monitoring disease progression, managing complications, and ensuring comprehensive treatment.
Case Report
A six-year-old male child, the third in birth order, born of a non consanguineous marriage, presented with facial puffiness and reduced urine output for four days following a recent upper respiratory tract infection two weeks prior. He had a history of high-grade fever, which was continuous in nature and associated with cold and cough. There were no other gastrointestinal symptoms. The antenatal history of the mother was uneventful. He was delivered full term by normal vaginal delivery, weighing 2.5 kg, and cried immediately after birth. He was diagnosed with congenital heart disease (ASD) and a short left upper limb. Family history was not significant, and the child was developmentally appropriate for his age. He attended school regularly and played well with his peers.
On examination, he was conscious, oriented, and febrile, with a temperature of 101°F. His blood pressure was 130/110 mmHg, which was above the 95th percentile. The respiratory rate was 30 breaths per minute. His weight on admission was 19 kg, and his height was 116 cm. His height-for-age was between the 50th and 90th percentiles, and his BMI was 14.12 kg/m2, indicating he was underweight. Pedal oedema and pallor were present. There was no icterus, cyanosis, clubbing, or lymphadenopathy. On cardiovascular examination, a second heart sound was heard with a wide fixed split, indicating ASD. On abdominal examination, there was distension with ascites, as shifting dullness was present. The rest of the systemic examination was normal. Haematological investigations and biochemical tests showed a complete blood count with mild anaemia (Haemoglobin of 9.8 g/dL) and leukocytosis (White blood cell count of 16,200 g/dL) with a normal platelet count of 281,000/μL. Blood sugar and serum electrolyte levels were normal. C-Reactive Protein (CRP) was elevated at 60 mg/dL. Urine examination revealed the presence of 15 to 20 red blood cells. Renal function tests were deranged, with serum creatinine increasing from 0.80 mg/dL at baseline to 1.37 mg/dL after three days. Serum urea levels increased from 36 mg/dL to 70 mg/dL. The urine protein-to-creatinine ratio was 1.1, and urinary protein was 3+. The Antistreptolysin O (ASO) titer was positive at 328 IU. C3 complement levels were decreased at 16.30 mg/dL (normal range: 90-180 mg/dL), while C4 complement levels were normal at 23 mg/dL (normal range: 10-40 mg/dL). Liver function tests were normal.
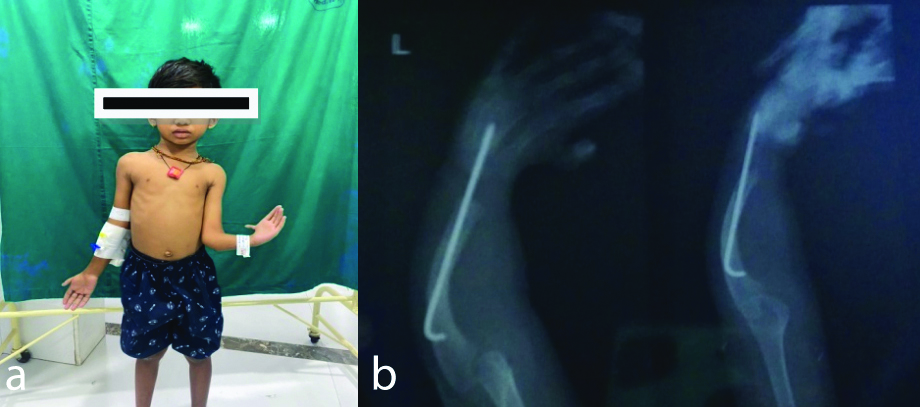
Radiological investigations showed a normal chest X-ray. The hand X-ray revealed a rod insertion for a radial abnormality and a K-wire for a finger procedure performed when the child was two years old [Table/Fig-1]. Ultrasonography of the abdomen and pelvis showed bilateral increased renal cortical echogenicity and mild ascites.
a) Classic deformity in the left upper forearm and hand. b) X-ray for the corrected deformity.
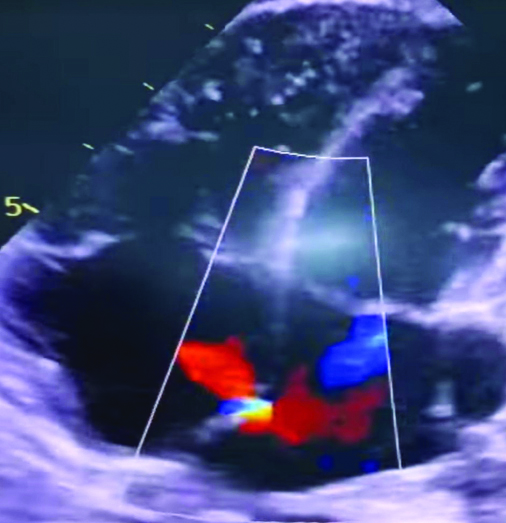
The 2D ECHO showed a 2 mm Atrial Septal Defect (ASD) [Table/Fig-2]. In view of the ASD, a cardiologist’s opinion was sought. As the defect was asymptomatic and small in sise, regular monitoring and yearly follow-up were advised.
A 2D ECHO shows: 2 mm Atrial Septal Defect.
Considering the history of a respiratory infection 15 days prior to presentation, along with facial puffiness, decreased urine output, haematuria, hypertension, oedema, ascites, a positive ASO titer, and deranged renal function with decreased C3 levels, a diagnosis of acute (PSGN) in a case of Holt-Oram syndrome was made. The diagnosis was based on the clinical background, and genetic testing could not be conducted due to financial constraints. Since there was no significant proteinuria, nephritic syndrome was ruled out, and the absence of pus cells in the urine ruled out a Urinary Tract Infection (UTI).
The child was admitted and placed in a propped-up position due to distress from ascites. Saturation on room air was normal. He was started on a course of intravenous antibiotics, ceftriaxone at 100 mg/kg, and due to hypertension, amlodipine was initiated at a dose of 5 mg, which was escalated to 7.5 mg once in the morning. The blood pressure was recorded at 130/110 mmHg, which was above the 95th percentile. A diuretic, furosemide, was added to achieve better control of hypertension. As blood pressure readings remained high, metoprolol 5 mg was added in the evening. The hospital stay was prolonged due to persistent fever spikes from sepsis, with CRP levels rising to 60 mg/dL from 18 mg/dL, alongside persistent hypertension. The antibiotic regimen was escalated to intravenous piperacillin and tazobactam at 100 mg/kg/day. Strict blood pressure monitoring was maintained, and the child clinically improved, with a blood pressure of 100/82 mmHg (50th to 90th percentile). He was discharged after three weeks of hospitalisation on amlodipine 7.5 mg once in the morning and metoprolol 5 mg in the evening. The parents were instructed on how to record blood pressure using an automated machine.
The child was followed-up after 15 days and one month, during which blood pressure remained elevated at both visits. After three months, the child was taken off the antihypertensive medication and was monitored again at 15-day and one-month intervals, during which blood pressure was found to be normal.
Discussion
Holt-Oram syndrome, also known as heart-hand syndrome or cardiomelic syndrome, is a congenital autosomal dominant disorder characterised by upper limb anomalies and congenital heart lesions due to mutations in the TBX5 gene. The incidence is approximately 1 in 100,000 live births, and it is distinguished by the absence of the radius bone in the upper extremities and ostium secundum type ASD [1-4]. The presence of these features in present case was suggestive of Holt-Oram syndrome.
Acute PSGN was diagnosed, as the child had a history of respiratory infection two weeks prior to presentation, accompanied by facial puffiness, decreased urine output, microscopic haematuria, hypertension, oedema, ascites, a positive ASO titer, deranged renal function, and decreased C3 levels. A differential diagnosis for Holt-Oram syndrome includes Thrombocytopenia Absent Radius (TAR) syndrome, which is characterised by thrombocytopenia. Since the child had a normal platelet count, TAR syndrome was ruled out.
TAR syndrome is a congenital disorder with an autosomal recessive pattern of inheritance, characterised by thrombocytopenia and bilateral absence or aplasia of the radii of the forearms. It can also be associated with skeletal, cardiac, renal, or gastrointestinal defects [5].
The cardiac abnormalities, such as heart blocks and arrhythmias, have been reported more commonly in Holt-Oram syndrome. Renal involvement has been rarely reported. Ferriols Gil EJ et al., documented a case of unilateral renal agenesis [6]. Patients with unilateral renal agenesis typically have a relatively good prognosis, provided that the remaining kidney is healthy and compensates for the missing one. However, they may be at a higher risk of developing hypertension, proteinuria, or chronic kidney disease later in life due to the increased workload on the solitary kidney. Dasgupta MK reported a case of a one-day-old baby with a unilateral hypoplastic kidney [7]. The prognosis for children with a hypoplastic kidney varies. If the remaining kidney functions well, they can have a normal life span. However, there is an increased risk of renal complications, similar to those seen with unilateral renal agenesis, including potential hypertension and chronic kidney disease. The management includes immediate neonatal care to assess the overall health of the baby and the functioning of the kidneys, as well as long-term monitoring with regular follow-ups with a paediatric nephrologist to evaluate renal function, growth, and development. Jain J et al., reported a case of a 17-year-old with renal agenesis in Holt-Oram syndrome [8]. The prognosis depends on the function of the remaining kidney. The patient may live normally but must be monitored for any signs of renal dysfunction. Regular renal function tests and blood pressure monitoring should be conducted. It is important to encourage practices that protect kidney function, such as avoiding excessive protein intake and nephrotoxic substances. Educating the patient about the signs of kidney problems and the importance of maintaining a healthy lifestyle is also crucial.
Ultrasound imaging in present case showed bilateral raised renal cortical echogenicity due to the associated PSGN. In the paediatric age group, persistent increases in blood pressure are commonly associated with chronic renal diseases. Most patients with PSGN experience complete clinical recovery, and the resolution of the disease process typically begins within the first two weeks. However, in present case, the blood pressure remained consistently high and required two antihypertensive medications to achieve control. This clinical presentation is very rare. A study by Bhalla K et al., reported the presence of persistent hypertension in a child with PSGN [9].
The child underwent rod insertion for radial abnormality and a K-wire procedure for finger issues at the age of two. He was advised to have physiotherapy to improve hand movement. Hafkemeyer U reported that interventions such as physiotherapy aid in patient rehabilitation and help prevent future complications [10]. The parents were counseled about the disease pattern and the importance of follow-up due to persistent hypertension. At the three-month follow-up, C3 levels had increased to normal, and blood pressure was controlled with a single antihypertensive, Amlodipine 5 mg.
Holt-Oram syndrome rarely presents with renal involvement, making this case unique. The association between Holt-Oram syndrome and acute glomerulonephritis underscores the need for comprehensive evaluation in such cases. Additionally, persistent hypertension in the context of acute glomerulonephritis is uncommon in paediatric patients, warranting close monitoring and management.
Conclusion(s)
Children with Holt-Oram syndrome may develop rare complications, such as acute glomerulonephritis, necessitating careful evaluation and management. Persistent hypertension in these cases may require multidrug therapy for control. Close follow-up is essential to monitor disease progression and ensure optimal outcomes. The co-occurrence of PSGN in a child with Holt-Oram syndrome could be coincidental, although the association cannot be ruled out.
[1]. Krauser AF, Ponnarasu S, Schury MP, Holt-Oram SyndromeIn: StatPearls [Internet] 2024 Treasure Island (FL)StatPearls PublishingAvailable from: https://www.ncbi.nlm.nih.gov/books/NBK513339/. [Updated 2023 Aug 14] [Google Scholar]
[2]. Nishijuka F, Suchen F, Sanches R, Holt oram syndrome case reportJ Am Coll Cardiol 2022 79(9_Supplement):3442 [Google Scholar]
[3]. Mori AD, Bruneau BG, TBX5 mutations and congenital heart disease: Holt-Oram syndrome revealedCurr Opin Cardiol 2004 19(3):211-15.10.1097/00001573-200405000-0000415096952 [Google Scholar] [CrossRef] [PubMed]
[4]. Wall LB, Piper SL, Habenicht R, Oishi SN, Esaki M, Goldfarb CA, Defining features of the upper extremity in Holt-Oram SyndromeJ Hand Surg Am 2015 40(9):1764-68. [Google Scholar]
[5]. Farlett R, Kulkarni A, Thomas B, Mydam J, Thrombocytopenia with absent radii syndrome with an unusual urological pathology: A case reportCureus 2022 14(4):e23991PMCID: PMC900187410.7759/cureus.2399135463560 [Google Scholar] [CrossRef] [PubMed]
[6]. Ferriols Gil EJ, Fayos Soler JL, Elorsa Arismendi J, Alvares Angel V, Holt-Oram syndrome. Presentation of two cases (author’s transl)Anales Espanoles de Pediatria 1981 15(4):378-82.7337303 [Google Scholar] [PubMed]
[7]. Dasgupta MK, Unilateral hypoplastic kidney in a case of Holt-Oram SyndromeJournal of Nepal Paediatric Society 2013 33(1):77-79. [Google Scholar]
[8]. Jain J, Janaarthanan S, Bondge B, Khan S, Joshi M, Banait T, Atypical Holt-Oram syndrome presenting with renal agenesis: A case reportRes-Int J Med Res Health Sci 2022 1(11):31-35. [Google Scholar]
[9]. Bhalla K, Gupta A, Nanda S, Mehra S, Epidemiology and clinical outcomes of acute glomerulonephritis in a teaching hospital in North IndiaJ Family Med Prim Care 2019 8(3):934-37.PMCID: PMC648274710.4103/jfmpc.jfmpc_57_1931041228 [Google Scholar] [CrossRef] [PubMed]
[10]. Hafkemeyer U, Holt-Oram syndrome. 3 case reports and their physical therapy, occupational therapy and technical orthopedic treatmentThe Orthopaedist 2001 30(4):226-30. [Google Scholar]