Huntington’s Disease (HD) is a rare neurodegenerative condition inherited in an autosomal dominant pattern, where Gamma Amino Butyric Acid-ergic (GABAergic) neurons in the basal ganglia progressively deteriorate. Symptoms include subcortical dementia, behavioural changes, midlife psychosis, and involuntary choreoathetosis movements. Imaging techniques like Computed Tomography (CT) and Magnetic Resonance Imaging (MRI) show shrinkage of the caudate nucleus and putamen, alongside enlarged lateral ventricles. Diagnosis is confirmed through molecular analysis identifying the hallmark amplified Cytosine Adenine Guanine (CAG) triplet. The case report describes a 57-year-old female diagnosed with HD, presenting symptoms of accidental falls caused by involuntary jerky movements, along with behavioural changes and cognitive decline over three years. Initial Non Contrast Computed Tomography (NCCT) of the brain revealed bilateral basal ganglia and diffuse cerebral atrophy. Quantitative analysis indicated a decreased frontal horn width to intercaudate distance (FH/CC) ratio of 1.24 and an increased Intercaudate distance to inner table width (CC/IT) ratio of 0.30. Multivoxel Magnetic Resonance Spectroscopy (MRS) showed reduced N-Acetyl Alanine (NAA) and creatine (Cr) levels with an NAA/Cr ratio of 0.84. Magnetic Resonance Tractography and Diffusion Tensor Imaging (DTI) revealed elevated Fractional Anisotropy (FA) and diffusivity in the basal ganglia, and reduced FA with increased diffusivity in white matter tracts. Genetic analysis by Polymerase Chain Reaction (PCR) confirmed HD. Newer MRI like DTI allows early identification of disease and better assessment of disease progression and response to therapy. A multidisciplinary approach involving neurologists, psychiatrists, and other healthcare professionals is essential. Genetic counseling and psychosocial support are crucial for patients and their families. Early and accurate identification of HD is vital for timely interventions and improved symptom control.
Case Report
A 57-year-old female presented with complaints of falls due to involuntary sporadic movements, along with shifts in behaviour and a decline in cognitive abilities over the last three years. These movements didn’t involve fixed postures. As per her husband, she had been healthy with no issues until three years ago. But now she has developed sudden involuntary jerky movements of her limbs that have been worsening lately. These movements intensify during stress or excitement but ease off during rest. Cognitive assessment revealed difficulties with attention, executive functions, and memory. There was no history of substance abuse in the past.
All her laboratory investigations were unremarkable. Initially, a Non Contrast Computed Tomography (NCCT) study of the brain revealed bilateral basal ganglia and diffuse cerebral atrophy [Table/Fig-1a,b].
a) NCCT of normal brain. Axial section at the level of basal ganglia (arrow). b) NCCT of brain in Huntington’s Disease (HD). Axial section at the level of basal ganglia showing bilateral corpus striatum atrophy (arrow).

Subsequently, an Magnetic Resonance Imaging (MRI) of the brain was performed, which demonstrated atrophy of the corpus striatum involving bilateral caudate nucleus and putamen, along with notable enlargement of the frontal horns of the lateral ventricles [Table/Fig-2,3].
a) MRI of normal brain. Axial section T2 Weighted Image (T2WI) at the level of basal ganglia (arrow). b) MRI of brain in Huntington’s Disease (HD). Axial section T2WI at the level of basal ganglia showing bilateral corpus striatum atrophy (arrow).

MRI of brain in Huntington’s Disease (HD) (Axial and coronal section T2WI and Fluid-attenuated Inversion Recovery (FLAIR) images showing atrophy of caudate nucleus with prominence of frontal horns of bilateral-lateral ventricles (arrows); also a,b) show increased intercaudate to inner table width (CC/IT) ratio and decreased frontal horn width to inner table width ratio.
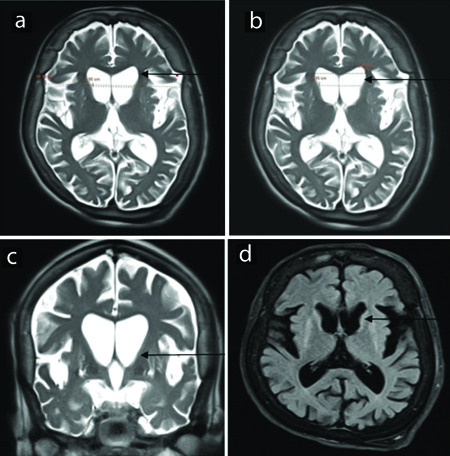
Quantitative analysis revealed a decreased frontal horn width/intercaudate distance ratio (FH/CC) of about 1.24 and an increased intercaudate distance to inner table width ratio (CC/IT) of about 0.30.
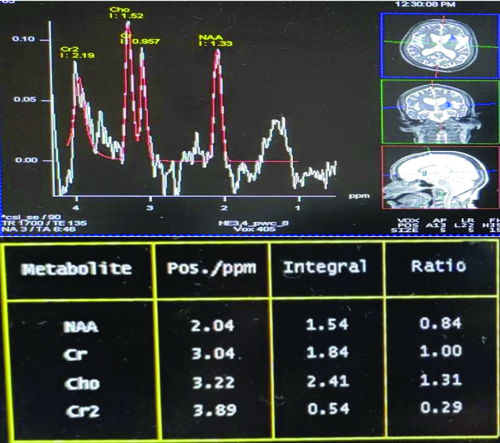
Multivoxel Magnetic Resonance Spectroscopy (MRS) depicted reduced NAA levels and creatine (Cr) levels in the bilateral basal ganglia, with an NAA/Cr ratio of 0.84 [Table/Fig-4].
MRS of the brain shows reduced NAA and creatine levels with increased choline levels.
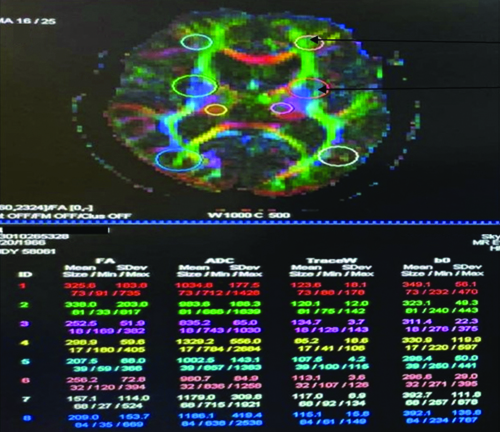
Magnetic Resonance Tractography and Diffusion Tensor Imaging (DTI) unveiled elevated Fractional Anisotropy (FA) and diffusivity in the bilateral basal ganglia, while demonstrating reduced FA and increased diffusivity in white matter tracts [Table/Fig-5].
DTI shows increased Fractional Anisotropy (FA) and increased Apparent Diffusion Coefficient (ADC) in bilateral basal ganglia and reduced FA and increased ADC in white matter tracts.
Further molecular genetic analysis by Polymerase Chain Reaction (PCR) for the Cytosine-Adenine-Guanine (CAG) trinucleotide in the Huntington disease Interesting Transcript 15 (IT15) gene showed about 44 CAG trinucleotide repeats, thus confirming the diagnosis.
The patient was referred to the neurology clinic for further management, where she was advised tablet tetrabenzine for control of her involuntary movements, initial dose at 12.5 mg/day once in morning for one week followed by twice daily for the next week and review in Outpatient Department (OPD). Later she did not review in the OPD.
Discussion
The HD, also referred to as Huntington’s chorea, is a rare and inherited neurodegenerative disorder, marked by the progressive loss of GABAergic neurons in the basal ganglia [1]. The condition affects approximately 1-7 individuals per million and is caused by the CAG repeat expansion on the p-arm of chromosome 4 [1,2]. HD is fully penetrant, meaning that individuals who inherit the defective gene will develop the disorder [3].
The choreatic form of HD is characterised by involuntary, jerky movements, whereas the Parkinsonian form involves slow movements and stiffness. Juvenile-onset HD begins in childhood or adolescence and advances rapidly, while late-onset HD develops later in life and often presents with milder symptoms [4]. Clinically, HD is associated with a blend of motor difficulties, cognitive decline, and emotional disturbances. Motor symptoms encompass involuntary jerky movements (chorea), persistent muscle contractions (dystonia), and problems with initiating or controlling movements [5]. Psychological symptoms such as irritability, anxiety, and sadness often precede motor symptoms. Other manifestations can include rigidity (Westphal variant), dementia, and emotional disturbances [6,7].
In HD, characteristic changes include degeneration of the basal ganglia, particularly the caudate and putamen regions, leading to their reduced size and absence of typical prominence [8]. Typical MRI findings in HD include atrophy of the caudate nucleus and heightened signal intensity on T2-weighted images in the basal ganglia and thalamus. The Anterior Commissure to Posterior Commissure (ACPC) line is used to assess the ratio between the caudate heads and the inner table of the skull. Normally, the CC/IT ratio ranges from 0.09 to 0.12, while the FH/CC ratio averages between 2.2 and 2.6. As the caudate heads atrophy, the distance between the caudate heads and the inner table of the skull increases, resulting in a higher CC/IT ratio [8-10].
An MRI study conducted by Aylward EH et al., revealed a strong correlation between caudate volume and both the FH/CC and CC/IT ratios in HD patients [11]. In another study, Mirowitz SA et al., reported bilateral symmetric T2 hyperintensity in the basal ganglia of two paediatric HD patients from a group of 65 children with neurodegenerative conditions [12]. Similarly, Telenius H et al., showed through their study, increased CAG repeat lengths and mosaicism not only in the brains but also in the sperms of individuals with HD [13]. Additionally, Pavese N et al., utilised raclopride imaging to reveal decreased cortical binding in a significant number of symptomatic HD subjects and premanifest carriers, with notable reductions observed in the temporal and frontal regions [14].
Radiological Differential Diagnosis for Caudate Atrophy
Caudate atrophy is a significant radiological finding indicative of several neurodegenerative and metabolic disorders. Neuroacanthocytosis syndromes encompass Chorea-Acanthocytosis (ChAc) and McLeod Syndrome characterised by movement disorders, psychiatric symptoms, and distinctive radiological features, such as caudate nucleus and putamen atrophy visible on MRI. Diagnosis involves identifying acanthocytes on blood smear and genetic testing [15]. Frontotemporal Lobar Degeneration (FTLD), specifically the variant with Fused in Sarcoma (FUS) protein, presents with behavioural changes and language difficulties. Radiologically, it manifests as caudate and frontal/temporal lobe atrophy, confirmed by histopathology showing FUS protein aggregation [16].
Wilson Disease manifests with hepatic, psychiatric, and neurological symptoms alongside MRI hyperintensities in basal ganglia and thalamus. Diagnosis is confirmed by low ceruloplasmin levels and genetic testing [16].
Leigh Disease, a mitochondrial disorder, shows symmetric T2 hyperintensities in basal ganglia and brainstem on MRI, diagnosed through genetic testing and elevated lactate levels in blood and Cerebrospinal Fluid (CSF) [17].
Pantothenate Kinase-associated Neurodegeneration (PKAN) exhibits dystonia, Parkinsonism, and cognitive decline with a characteristic “eye of the tiger” sign on MRI, indicating globus pallidus involvement, confirmed by genetic testing for Pantothenate Kinase 2 (PANK2) mutations [18]. Accurate diagnosis of caudate atrophy involves correlating clinical symptoms with specific radiological findings and confirming through laboratory tests and genetic analyses. Recognising distinct imaging patterns and associated clinical features is essential for effective diagnosis and management.
The present case report highlights several unique aspects of HD. It illustrates the classic clinical progression of involuntary jerky movements (chorea), worsening under stress, alongside cognitive decline. Radiologically, it demonstrates specific features such as bilateral atrophy of the caudate nucleus and putamen, enlarged frontal horns of the lateral ventricles, and quantitative MRI markers like the FH/CC and CC/IT ratios. The molecular genetic analysis confirmed the diagnosis with 44 CAG trinucleotide repeats, emphasising the role of genetic testing in definitive diagnosis and management decisions. Given its familial nature, HD also profoundly impacts family members. As HD is genetic, ethical considerations regarding predictive testing, family planning, and genetic information disclosure are pertinent. Healthcare professionals must approach these complex ethical dilemmas with sensitivity and empathy.
Conclusion(s)
The HD has wide-ranging effects on a person’s life, affecting motor function, cognition, and mental health. As any definitive treatment of HD is yet to be developed, a comprehensive approach to care, involving neurologists, psychiatrists, physical therapists, and social workers is crucial. MRI and its recent advancements have an immense role in diagnosing and assessing the severity of the disease. Newer MRI like DTI allows early identification of the disease in the premanifest stage and allows better assessment of disease progression and response to therapy.
[1]. Reiner A, Dragatsis I, Dietrich P, Genetics and neuropathology of Huntington’s diseaseInt Rev Neurobiol 2011 98:325-72. [Google Scholar]
[2]. Nopoulos PC, Huntington disease: A single-gene degenerative disorder of the striatumDialogues Clin Neurosci 2016 18:91-98. [Google Scholar]
[3]. Lee JM, Ramos EM, Lee JH, Gillis T, Mysore JS, Hayden MR, CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashionNeurology 2012 78:690-95. [Google Scholar]
[4]. Quarrell OW, Nance MA, Nopoulos P, Paulsen JS, Smith JA, Squitieri F, Managing juvenile Huntington’s diseaseNeurodegener Dis Manag 2013 3(3):267-76. [Google Scholar]
[5]. Andhale R, Shrivastava D, Huntington’s disease: A clinical reviewCureus 2022 14:e28484 [Google Scholar]
[6]. Jevtic SD, Provias JP, Case report and literature review of Huntington disease with intermediate CAG expansionBMJ Neurol Open 2020 2:e000027 [Google Scholar]
[7]. Gonzalez-Alegre P, Afifi AK, Clinical characteristics of childhood-onset (juvenile) Huntington disease: Report of 12 patients and review of the literatureJ Child Neurol 2006 21:223-29. [Google Scholar]
[8]. Van Cauter S, Severino M, Ammendola R, Van Berkel B, Vavro H, van den Hauwe L, Bilateral lesions of the basal ganglia and thalami (central grey matter)-pictorial reviewNeuroradiology 2020 62:1565-605. [Google Scholar]
[9]. Negi RS, Manchanda KL, Sanga S, Imaging of Huntington’s diseaseMed J Armed Forces India 2014 70:386-88. [Google Scholar]
[10]. Deng JH, Zhang HW, Liu XL, Deng HZ, Lin F, Morphological changes in Parkinson’s disease based on magnetic resonance imaging: A mini-review of subcortical structures segmentation and shape analysisWorld J Psychiatry 2022 12:1356-66. [Google Scholar]
[11]. Aylward EH, Schwartz J, Machlin S, Pearlson G, Bicaudate ratio as a measure of caudate volume on MR imagesAm J Neuroradiol 1991 12:1217-22. [Google Scholar]
[12]. Mirowitz SA, Sartor K, Prensky AJ, Gado M, Hodges FJ 3rd, Neurodegenerative diseases of childhood: MR and CT evaluationJ Comput Assist Tomogr 1991 15:210-22. [Google Scholar]
[13]. Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in brain and spermNat Genet 1994 6:409-14. [Google Scholar]
[14]. Pavese N, Politis M, Tai YF, Barker RA, Tabrizi SJ, Mason SL, Cortical dopamine dysfunction in symptomatic and premanifest Huntington’s disease gene carriersNeurobiol Dis 2010 37:356-61. [Google Scholar]
[15]. Walker RH, Jung HH, Danek A, Rubio JP, Auburger G, Neuroacanthocytosis syndromesOrphanet J Rare Dis 2007 2:4 [Google Scholar]
[16]. Dusek P, Litwin T, Czlonkowska A, Wilson disease and other neurodegenerations with metal accumulationsNeurol Clin 2015 33(1):175-204. [Google Scholar]
[17]. Sofou K, De Coo IF, Isohanni P, Ostergaard E, Naess K, De Meirleir L, A multicenter study on Leigh syndrome: Disease course and predictors of survivalOrphanet J Rare Dis 2014 9:52 [Google Scholar]
[18]. Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ, A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndromeNat Genet 2001 28(4):345-49. [Google Scholar]