A Novel Methyl-CpG Binding Protein 2 (MeCP2) Variant in an Indian Girl with Rett Syndrome
Pratiksha Chheda1, Shailesh Pande2, Tavisha Dama3, Dollar Goradia4, Sushant Vinarkar5
1 Senior Manager, Department of Molecular Pathology, Metropolis Healthcare Ltd., Mumbai, Maharashtra, India.
2 Head, Department of Medical Genetics, Metropolis Healthcare Ltd., Mumbai, Maharashtra, India.
3 Scientific Officer, Department of Molecular Pathology, Metropolis Healthcare Ltd., Mumbai, Maharashtra, India.
4 Scientific Officer, Department of Molecular Pathology, Metropolis Healthcare Ltd., Mumbai, Maharashtra, India.
5 Head, Department of Molecular Pathology, Metropolis Healthcare Ltd., Mumbai, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Dr. Pratiksha Chheda, Metropolis Healthcare Ltd., Unit No. 409 to 416, 4th Floor, Commercial Building A, Kohinoor City, Near Kohinoor Mall, Kirol Road, Kurla-W, Mumbai-400070, Maharashtra, India.
E-mail: pratikshad13@gmail.com
Rett syndrome is an X-linked dominant disorder that is primarily seen in females and is linked to mutations in the gene coding for Methyl-CpG Binding Protein 2 (MeCP2). It is a neurodevelopmental disorder characterised by impairments in language, repetitive movements, early-onset seizures, delayed growth, autistic features, intellectual disability and abnormal Electroencephalograms (EEG). Author’s reported a case of three year six months old Indian girl who was born of a nonconsanguineous marriage presented with stereotypic hand movements, gradual loss of speech, inability to walk independently and frequent episodes of seizure. Genetic testing for analysis of MeCP2 mutations was performed and a novel de novo missense variant (c.361G>A, p.Asp121Asn) was identified, which was predicted to be disease causing on the basis of insilico analysis and clinical findings. The study suggested that a careful evaluation of the pathogenic nature of MeCP2 variants supports clinical diagnosis and aids in genetic counseling and patient management.
Epileptiform discharge, Mutations, Neurological syndrome, Seizures, X-linked dominant
Case Report
A three year six months old female child presented to the children’s hospital with complaints of breathing difficulties and had seizures since one month. She was born out of a nonconsanguineous marriage, following an uncomplicated pregnancy and full term normal delivery. The mother reported that the child was healthy, cried immediately and fed well. Normal development was observed until the age of 12 months, following which there was a gradual loss of speech, and by the age of three years it was restricted to mere babbling. She was unable to walk independently and could walk slowly with support. Mother was apparently normal and none other family members were affected with similar condition. Stereotypic hand movement included repeated movement of hand towards the mouth, specifically constant biting of middle and index fingers. Skeletal abnormality such as scoliosis was not present; however, she had genu valgum. She showed no interest in playing with toys. Head circumference was normal. She used to have disturbed sleep pattern, but it had improved with medication. She had difficulty in breathing.
At the age of three years and three months seizure-like episodes that lasted for one minute were observed once in every 2-3 days. The case was further evaluated and a series of investigations were carried out. MRI and NonContrast CT (NCCT) brain were performed and no abnormalities were identified. Further EEG was done using standard 10-20 international electrode placement systems while patient was awake first, and later asleep. EEG findings were recorded as abnormal in view of: i) epileptiform discharges; ii) very frequent generalised bursts followed by mild electrodecremental response; and iii) generalised paroxysmal fast activity. The patient was put on treatment for seizures and had shown signs of improvement. Rett syndrome was suspected based on clinical presentation and EEG findings and patient was referred to us for genetic evaluation of MeCP2 gene.
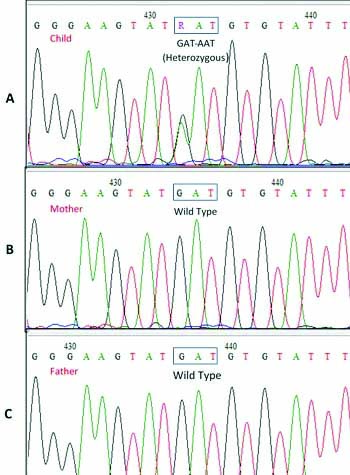
The genomic DNA was extracted from patient’s peripheral blood (200 μL) using the Qiagen DNA Mini kit. Exons 1-4 of MeCP2 gene were amplified using primers and method described earlier [1]. The primer sequences and corresponding annealing temperature and amplimer sizes are given in [Table/Fig-1]. The purified PCR products were sequenced directly using the BigDye® Terminator version 3.1 Cycle Sequencing Kit (Thermo Fisher Scientific) on Applied Biosystems 3500 Dx Genetic Analyser. The sequences were aligned to the reference sequence available in NCBI database (Genbank accession no.: NM_004992.3) using the Bioedit software. A single base change – c.361G>A, p.Asp121Asn in exon 3 of MeCP2 gene was identified [Table/Fig-2]. This variant was not reported previously in ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/), RettBASE: Rett Syndrome Variation Database (mecp2chw.edu.au/) or 1000 Genome database, nor was it found in the published literature [2]. The insilico analysis using PolyPhen-2, MutationTaster and Sifting Intolerant From Tolerant (SIFT) predicted this novel variant to be damaging or pathogenic. In order to establish de novo nature of the variant, parental blood samples were analysed for presence of c.361G>A variant. Sequencing of exon 3 of MeCP2 gene showed absence of c.361G>A variant in both the parents [Table/Fig-2]. Informed consent was obtained from the parents for genetic testing. The parents were counseled for the condition and patient was asked to meet the referring doctor for further management. The report was discussed with the referring doctor and the importance of regular medical care including medication for associated symptoms such as seizures, muscle stiffness, heart and gastrointestinal issues etc. Also, need for physical, occupational, nutritional, speech and language therapy was discussed with the parents.
MeCP2: Primer sequences, Annealing temperatures and Amplimer sizes.
| Primer | Sequence | Annealing Temperature | Amplimer Size (bp) |
|---|
| Exon 1-F | 5′–GCACTCGGTGCATCTGTGGACAGAG–3′ | 65°C | 671 |
| Exon 1-R | 5′–CATCCGCCAGCCGTGTCGTCCGAC–3′ |
| Exon 2-F | 5′–TGTGTTTATCTTCAAAATGT–3′ | 57°C | 418 |
| Exon 2-R | 5′–GTTATGTCTTTAGTCTTTGG–3′ |
| Exon 3-F | 5′–CTGGGGCCTTGCATGTGGTG–3′ | 58°C | 626 |
| Exon 3-R | 5′–GGTCATTTCAAGCACACCTG–3′ |
| Exon 4a1-F | 5′–CGAGTGAGTGGCTTTGGTGA–3′ | 60°C | 305 |
| Exon 4a1-R | 5′–ACAGATCGGATAGAAGACTC–3′ |
| Exon 4a2-F | 5′–CCACCCAGGTCATGGTGATC–3′ | 58°C | 384 |
| Exon 4a2-R | 5′–TGAGTGGTGGTGATGGTGGT–3′ |
| Exon 4b-F | 5′–GCAGGAGACCGTACTCCCCATC–3′ | 66°C | 366 |
| Exon 4b-R | 5′–GCTCTCCCTCCCCTCGGTGT–3′ |
| Exon 4c/d-F | 5′–GCAGGAGACCGTACTCCCCATC–3′ | 63°C | 375 |
| Exon 4c/d-R | 5′–GCTCTCCCTCCCCTCGGTGT–3′ |
| Exon 4e-F | 5′–GGAGAAGATGCCCAGAGGAG–3′ | 60°C | 455 |
| Exon 4e-R | 5′–CGGTAAGAAAAACATCCCCA–3′ |
Discussion
Rett syndrome is an X-linked dominant neurodevelopmental disorder with an incidence rate of 1 in 10000–15000. It is one of the most common causes of intellectual disability in females and was originally described in the 1960’s by Andreas Rett [3,4]. Clinical diagnosis of Rtt rests on a careful exploration of clinical symptoms and specific pattern of symptom progression. The age of disease onset vary from later infancy with global developmental delays, gradual speech and communication impairments, progressive microcephaly, seizures and autistic behaviors. There is significant overlap in phenotypic features as well as disease severity which sometimes mask underlying diagnosis. Mutations in the gene encoding MeCP2 are present in 95-97% of patients with typical Rtt but only in 50-70% of atypical cases [5]. Majority (95%) of the Rtt cases are sporadic, show presence of de novo mutations in the MeCP2 gene that is not inherited from either parent and are mostly detected in females since affected male fetuses have lethal phenotype [6,7]. Over the last few years mutations in Cyclin Dependant Kinase Like 5 (CDKL5) and Forkhead Box Protein G1 (FOXG1) genes have also been documented to be associated with Rtt [5]. It is important that Rtt should be diagnosed and differentiated from other disorders with overlapping phenotype such as cerebral palsy and autism, as the patient management and outcomes differ significantly [8].
In the present case, the major clinical features comprised of stereotypic hand movements, gradual loss of speech, inability to walk independently and frequent episodes of seizure seizures. A missense variant (c.361G>A, p.Asp121Asn) was identified, which was not reported previously in any of the databases, however, insilico analysis predicted it to be damaging. It was not identified in the parents and segregation analysis confirmed de novo nature of the variant. In view of the clinical history provided and parental analysis, this de novo missense variant (c.361G>A, p.Asp121Asn) was classified as a ‘Likely Pathogenic’ (Class 4) mutation as per American College of Medical Genetics and Genomics (ACMG) variant classification guidelines, thereby confirming diagnosis of Rtt in the patient [9]. Parents were counseled regarding the implication of de novo mutation and disease risk to other family members.
Though close to 1000 different mutations in MeCP2 gene are recorded in the RETT database, there are certain mutations such as T158M, R168X, R255X, R270X and R306C found at higher frequencies [10-14]. Novel mutations comprising of missense, small insertions and deletions as well as large and complex rearrangements are being regularly identified [10,12,14]. Genetic evaluation of suspected Rtt cases thus involves complete MeCP2 gene sequencing, followed by analysis of large rearrangements in MeCP2 and testing of other associated genes if indicated. Clinical evaluation and genetic testing followed by counseling is important as early identification and timely intervention can help both the patient and their families.
Conclusion(s)
Diagnosis of Rtt is challenging due to diverse phenotypic variation. Careful evaluation of novel genetic variants along with genetic studies on parents to find de novo nature of the variant is important to establish genetic diagnosis.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Nov 06, 2020
Manual Googling: Jan 23, 2021
iThenticate Software: Mar 22, 2021 (20%)
[1]. Dumitriu S, Klootwijk E, Issler N, Stanescu H, Kleta R, Puiu M, Mutation analysis of the MECP2 gene in Romanian females with Rett syndromeRevista Româna de Medicina de Laborator 2013 21(4):437-45.10.2478/rrlm-2013-0038 [Google Scholar] [CrossRef]
[2]. Krishnaraj R, Ho G, Christodoulou J, RettBASE: Rett syndrome database updateHum Mutat 2017 38(8):922-31.10.1002/humu.2326328544139 [Google Scholar] [CrossRef] [PubMed]
[3]. Rett A, On an unusual brain atrophy syndrome in hyperammonemia in childhoodWien Med Wochenschr 1966 116(37):723-26. [Google Scholar]
[4]. Lee EY, Chung HJ, Ki CS, Yoo JH, Choi JR, A Novel Mutation in the MECP2 Gene in a Korean Patient with Rett SyndromeAnn Clin Lab Sci 2011 41(1):93-96. [Google Scholar]
[5]. Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, RettSearch Consortium. Rett syndrome: Revised diagnostic criteria and nomenclatureAnn Neurol 2010 68(6):944-50.10.1002/ana.2212421154482 [Google Scholar] [CrossRef] [PubMed]
[6]. Trappe R, Laccone F, Cobilanschi J, Meins M, Huppke P, Hanefeld F, MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal originAm J Hum Genet 2001 68(5):1093-101.10.1086/32010911309679 [Google Scholar] [CrossRef] [PubMed]
[7]. Filosa S, Pecorelli A, D’Esposito M, Valacchi G, Hajek J, Exploring the possible link between MeCP2 and oxidative stress in Rett syndromeFree Radic Biol Med 2015 88(Pt A):81-90.10.1016/j.freeradbiomed.2015.04.01925960047 [Google Scholar] [CrossRef] [PubMed]
[8]. Kumar S, Alexander M, Gnanamuthu C, Recent experience with Rett syndrome at a tertiary care centerNeurol India 2004 52(4):494-95. [Google Scholar]
[9]. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular PathologyGenet Med 2015 17:405-24.10.1038/gim.2015.3025741868 [Google Scholar] [CrossRef] [PubMed]
[10]. Lallar M, Rai A, Srivastava P, Mandal K, Gupta N, Kabra M, Molecular testing of MECP2 Gene in Rett syndrome phenotypes in Indian girlsIndian Pediatr 2018 55(6):474-77.10.1007/s13312-018-1336-y29428920 [Google Scholar] [CrossRef] [PubMed]
[11]. Bienvenu T, Villard L, De Roux N, Bourdon V, Fontes M, Beldjord C, Spectrum of MECP2 mutations in Rett syndromeGenet Test 2002 6(1):01-06.10.1089/10906570276009384312180070 [Google Scholar] [CrossRef] [PubMed]
[12]. Le Thi Thanh H, Do Thi Diem T, Duy CV, Thanh HLT, Phuong HBT, Thanh LN, Spectrum of MECP2 mutations in Vietnamese patients with RETT syndromeBMC Med Genet 2018 19(1):13710.1186/s12881-018-0658-x30081849 [Google Scholar] [CrossRef] [PubMed]
[13]. Philippe C, Villard L, De Roux N, Raynaud M, Bonnefond JP, Pasquier L, Spectrum and distribution of MECP2 mutations in 424 Rett syndrome patients: A molecular updateEur J Med Genet 2006 49(1):09-18.10.1016/j.ejmg.2005.04.00316473305 [Google Scholar] [CrossRef] [PubMed]
[14]. Maortua H, Martinez-Bouzas C, Garcia-Ribes A, Martinez MJ, Guillen E, Domingo MR, MECP2 gene study in a large cohort: Testing of 240 female patients and 861 healthy controls (519 females and 342 males)J Mol Diagn 2013 15(5):723-29.10.1016/j.jmoldx.2013.05.00223810759 [Google Scholar] [CrossRef] [PubMed]