Haemoglobin Olympia {β Codon 20 (B2) G→A, Val→Met}: A Silent Haemoglobin Variant
Abhay A Bhave1, Lakshmi Iyer2, Nawal Kazi3, Manju Gorivale4, Anita Nadkarni5
1 Hematologist, Department of Haematology, Empire Centre Haematology and Oncology Specialty Clinic, Mumbai, Maharashtra, India.
2 Clinical Assistant, Department of Haematology, Empire Centre Haematology and Oncology Specialty Clinic, Mumbai, Maharashtra, India.
3 Clinical Assistant, Department of Haematology, Empire Centre Haematology and Oncology Specialty Clinic, Mumbai, Maharashtra, India.
4 Laboratory Technician, Department of Haematology, National Institute of Immunohaematology (ICMR), Mumbai, Maharashtra, India.
5 Deputy Director, Department of Haematology, National Institute of Immunohaematology (ICMR), Mumbai, Maharashtra, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Anita Nadkarni, Deputy Director, Department of Haematology, National Institute of Immunoh aematology (ICMR), 13th Floor, New Multistoried Building, K.E.M. Hospital Campus, Parel, Maharashtra-400012, Mumbai, India.
E-mail: anitahnadkarni@yahoo.com
High oxygen affinity haemoglobin variants are rare and often underdiagnosed in persistent erythrocytosis with no apparent aetiology. Here the author present a 29-year-old Indian male patient with a long-standing history of erythrocytosis which was incidentally detected. The proband had a prothrombotic family history of cerebral vessel stroke in his paternal grandfather at a young age and unexplained erythrocytosis in his father and brother. A review of his haemograms showed persistent high haemoglobin values. Routine tests did not reveal any specific aetiology and haemoglobin electrophoresis by High-Performance Liquid Chromatography (HPLC) showed absence of any abnormal peak or unstable haemoglobin. DNA sequencing of the β globin gene revealed heterozygosity for codon 20 {GTG→ATG, Valine (Val)→ Methionine (Met)} mutation confirming the presence of an electrophoretically silent Hb variant - Haemoglobin Olympia in him and his extended family members. This case study emphasises importance of this rare entity of high oxygen affinity haemoglobin variant as a differential diagnosis while screening for erythrocytosis. This is the first case report of Haemoglobin Olympia from India reported in the literature.
Electrophoresis, Haematocrit, Mutation, Oxygen affinity, Polycythemia
Case Report
The proband is a 29-year-old Indian male patient, an ex-smoker with no known co-morbidities. In 2013, he was incidentally detected to have erythrocytosis on a routine blood evaluation. He did an initial workup for the cause of erythrocytosis and underwent phlebotomy infrequently. He then presented in the outpatient clinic in 2017 for the assessment of persistent erythrocytosis. He was asymptomatic with no past history of thrombosis. There was a family history of thrombosis (cerebral vascular accident) in his paternal grandfather in his young age and erythrocytosis in his brother and father. On clinical examination, the patient was plethoric with an SPO2 level normal (99%). He had no cyanosis, clubbing or organomegaly. His cardiac and pulmonary status was normal. Patient had well-established erythrocytosis {Red Blood Cell (RBC) 6.4×1012/L, Packed Cell Volume (PCV) 0.52 L/L, Mean Corpuscular Volume (MCV) 79 fL} with normal White Blood Cell (WBC) count and platelets. Peripheral smear showed hypochromia and anisocytosis. [Table/Fig-1] shows the periodic values of patient’s haemoglobin (Hb) and haematocrit levels.
Haemoglobin (Hb) and Packed Cell Volume (PCV) levels recorded periodically over a span of eight years.
| Parameters | 9 Sep 13 | 24 May 14 | 29 Dec 15 | 14 Dec 16 | 24 Jan 17 | 23 Feb 17 | 12 July 17 | 18 Jan 18 | 14 June 18 | 17 Oct 18 | 02 May 19 | 31 Aug 19 | 20 Dec 19 | 11 June 20 |
|---|
| Hb (gm %) | 17.9 | 18.8 | 16.5 | 16.3 | 16.3 | 16.8 | 16.1 | 15.5 | 15.7 | 15.9 | 15.7 | 15.5 | 15.6 | 16.3 |
| PCV (L/L) | 0.50 | 0.56 | 0.53 | 0.51 | 0.54 | 0.50 | 0.53 | 0.49 | 0.49 | 0.47 | 0.48 | 0.48 | 0.47 | 0.52 |
Biochemical investigations including renal and liver function tests were normal; Cholesterol was 5.24 mmol/L (Desirable level <5.17 mmol/L), Triglycerides were 1.77 mmol/L {Normal Range (NR): 0.11-2.15 mmol/L}, Lactate Dehydrogenase (LDH) was 263 U/L (NR: 50-200 U/L). Serum Ferritin levels were normal at 121 μg/L {NR: 4.63 to 204 μg/L by Chemiluminescent Immunoassay}. Serum Erythropoietin level was 9.85 IU/L {NR: 3.7 to 36 IU/L by Chemiluminescent immunoassay}. Leukocyte Alkaline Phosphatase (LAP) level tested by Cytochemistry was also within the normal range. No abnormalities were detected on Echocardiogram, Abdominal Ultrasound, Sleep study and Pulmonary Function Test.
Mutations for myeloproliferative neoplasm {Janus Kinase2 (JAK2) V617F mutation, JAK 2 exon 12 and MPL W515/S505 mutation} were negative by real time Polymerase Chain Reaction (PCR).
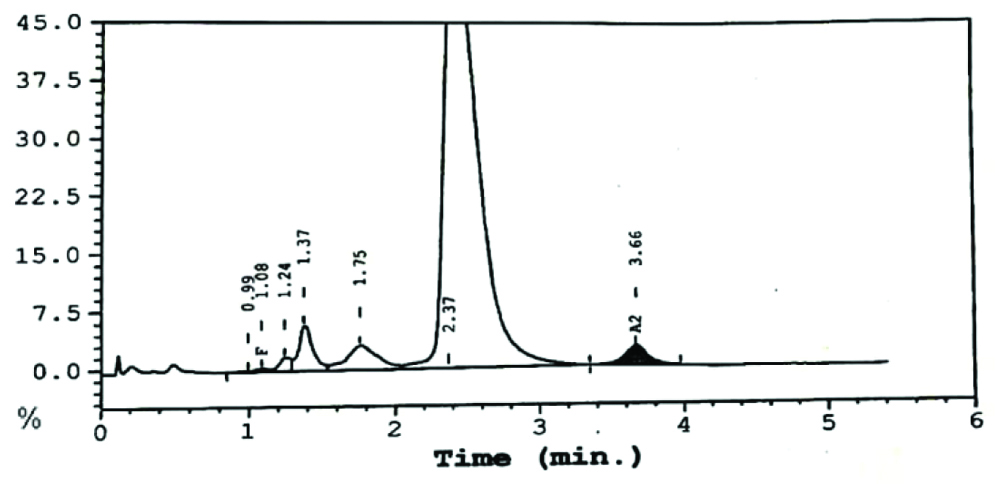
High Performance Liquid Chromatography (HPLC) done by the variant haemoglobin testing system (Bio-Rad Clinical Data Management (CDM) System) [Table/Fig-2] indicated no evidence of beta thalassaemia or haemoglobinopathy. Haemoglobin (Hb) F concentration was computed to be 0.3% and HbA2 was 2.5%. Heat stability test was found to be negative. P50 values to determine oxygen affinity were not done for this patient.
HPLC analysis on the Variant Hb Testing System (Bio-Rad CDM System).
HPLC: High performance liquid chromatography; Hb: Haemoglobin; CDM: Clinical data management
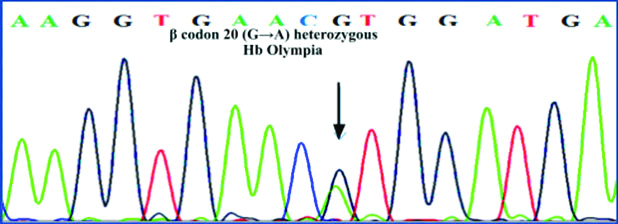
DNA sequencing of the β globin gene revealed heterozygosity for a mutation at codon 20 {GTG>ATG, VAL>Met} confirming the presence of an electrophoretically silent haemoglobin variant- Haemoglobin Olympia [Table/Fig-3].
Automated DNA Sequencing Electropherogram. β globin gene showing presence of codon 20 (GTG→ATG) Val →Met heterozygous mutation (Hb - Olympia trait).
Extended family study revealed presence of Haemoglobin Olympia in the proband’s father and younger brother.
Patient was advised to undergo therapeutic phlebotomy at regular intervals (if his Hb level increased to >15 gm% and PCV to >0.45 L/L) and to consume plenty of oral fluids with monitoring of Complete Blood Count (CBC). He was initiated on low dose aspirin (75 mg) to be taken once every day on a long-term basis. He was also counseled on the hazards of smoking and its consequences related to his condition. The procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation.
Discussion
Haemoglobinopathies are single gene disorders which affect the structure or synthesis of haemoglobin [1]. Thalassaemia arises when there is a quantitative defect in the globin chain while a structural change or change in quality of haemoglobin causes abnormal variants with altered physical properties, such as high oxygen affinity Hb variants that lead to erythrocytosis.
According to HbVar, a database of Human Haemoglobin Variants and Thalassaemias, over 100 haemoglobin variants exist which have increased affinity for oxygen [2].
The World Health Organisation (WHO) estimates that approximately 5.2% of the world’s population carries a notable haemoglobin variant [3]. Haemoglobinopathies are mainly found in people of Mediterranean, African, South-East Asian and Middle Eastern descent with varying clinical consequences [4]. Data from India suggests that the incidence of haemoglobinopathies is approximately 4.2% [5] and is known to contribute to morbidity and mortality.
The clinical presentation of high oxygen affinity Hb variants varies widely from being asymptomatic to showing symptoms like dizziness, fatigue, shortness of breath, easy bruising, vertigo, tinnitus, epistaxis, insomnia, paresthaesia in the limbs, hypertension, frequent headaches, facial redness, erythema and pruritus [6] with an additional risk of hyperviscosity syndrome.
Haemoglobin Olympia first described by Stamatoyannopoulos G et al., in 1973 occurs due to substitution of Valine by Methionine at beta globin gene codon 20 (B2) [7]. This is a high oxygen affinity hemoglobin variant causing erythrocytosis due to tissue hypoxia as a result of left shift of oxygen dissociation curve [8]. Erythropoietin (EPO) is an important hormone secreted by the kidney and the erythroid progenitor cells. In healthy patients, EPO levels vary inversely with haematocrit. When there is significant hypoxia at the cellular level, EPO is released by the kidney and this stimulates erythrocyte production. Abnormal oxygen sensing leads to increased production of EPO and hence increased haematocrit [9].
In literature, there have been few case reports of Hb Olympia, mainly from the United States and Europe [10-12] one of which is a Greek male having a combination of three molecular defects {Hb Olympia HBB:c.61G>A}, β thalassaemia {Haemoglobin Subunit Beta (HBB):c.118C>T} and α thalassaemia {αα /-- MED} which were detected together [13]. The patient in this study did not exhibit associated β or α thalassaemia which may be the reason for his milder phenotype. We performed the variant analysis in this patient as there was a history of erythrocytosis in his father and grandfather. This Hb variant is inherited in an autosomal dominant manner with an associated family history.
Treatment is mainly to prevent complications due to increased red cell mass and hence regular therapeutic phlebotomy and oral aspirin are advised so as to reduce the risk of complications like myocardial infarction, ischaemic stroke, or arterial or venous thromboembolism [14]. Target Hb/haematocrit level should be individualised as per patient’s clinical condition [15].
Conclusion(s)
Haemoglobin Olympia is a rare high oxygen affinity haemoglobin variant that causes erythrocytosis. Early detection and proper management can reduce morbidity and improve quality of life especially preventing young thromboembolic events as also avoiding exposure to drugs that can cause significant side-effects. Electrophoresis by HPLC and subsequent DNA sequencing play a pivotal role in the diagnosis. In a family with erythrocytosis, when electrophoretic and chromatographic studies fail to demonstrate a haemoglobin variant, the study of oxygen dissociation curves, P50 of affected individuals will be helpful in diagnosis. Characterisation of the variant haemoglobin needs to be done by PCR and Direct DNA sequencing.
Treatment with judicious therapeutic phlebotomies, hydration and oral antiplatelet agents along with regular blood count monitoring is recommended. This case study emphasises the importance of keeping this rare high oxygen affinity hemoglobin variant in the differential diagnoses of erythrocytosis across all age groups.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. NA
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Nov 03, 2020
Manual Googling: Dec 31, 2020
iThenticate Software: Feb 06, 2021 (3%)
[1]. Weatherall DJ, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P, Inherited disorders of hemoglobin. In: Disease Control Priorities in Developing Countries. 2nd edition. Jamison D et al. (Eds) 2006 Oxford University Press and the World Bank:663-680.10.1596/978-0-8213-6179-5/Chpt-34 [Google Scholar] [CrossRef]
[2]. HbVar: A Database of Human Hemoglobin Variants and Thalassemias. Globin gene Server home page. http://globin.cse.psu.edu [Google Scholar]
[3]. Modell B, Darlison M, Global epidemiology of haemoglobin disorders and derived service indicatorsBull World Health Organ 2008 86(6):480-87.10.2471/BLT.06.03667318568278 [Google Scholar] [CrossRef] [PubMed]
[4]. Weatherall DJ, The inherited diseases of hemoglobin are an emerging global health burdenBlood 2010 115(22):4331-36.10.1182/blood-2010-01-25134820233970 [Google Scholar] [CrossRef] [PubMed]
[5]. Colah R, Gorakshakar A, Nadkarni A, Phanasgaonkar S, Surve R, Sawant P, Regional heterogeneity of beta-thalassemia mutations in the multi ethnic Indian populationBlood Cells Mol Dis 2009 42(3):241-46.10.1016/j.bcmd.2008.12.00619254853 [Google Scholar] [CrossRef] [PubMed]
[6]. Yudin J, Verhovsek M, How we diagnose and manage altered oxygen affinity hemoglobin variantsAm J Hematol 2019 94(5):597-603.10.1002/ajh.2542530690774 [Google Scholar] [CrossRef] [PubMed]
[7]. Stamatoyannopoulos G, Nute PE, Adamson JW, Bellingham AJ, Funk D, Hemoglobin olympia (20 valine leads to methionine): An electrophoretically silent variant associated with high oxygen affinity and erythrocytosisJ Clin Invest 1973 52(2):342-49.10.1172/JCI1071904683875 [Google Scholar] [CrossRef] [PubMed]
[8]. Percy MJ, Butt NN, Crotty GM, Drummond MW, Harrison C, Jones GL, Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosisHaematologica 2009 94(9):1321-22.10.3324/haematol.2009.00803719734427 [Google Scholar] [CrossRef] [PubMed]
[9]. Haider MZ, Anwer F, Secondary Polycythemia 2020 Stat Pearls. Stat Pearls Publishing [Google Scholar]
[10]. Weaver GA, Rahbar S, Ellsworth CA, de Alarcon PA, Forbes GB, Beutler E, Iron overload in three generations of a family with hemoglobin OlympiaGastroenterology 1984 87(3):695-702.10.1016/0016-5085(84)90545-6 [Google Scholar] [CrossRef]
[11]. Percy MJ, Butt NN, Crotty GM, Drummond MW, Harrison C, Jones GL, Identification of high oxygen affinity hemoglobin variants in the investigation of patients with erythrocytosisHaematologica 2009 94(9):1321-1322.10.3324/haematol.2009.00803719734427 [Google Scholar] [CrossRef] [PubMed]
[12]. Esparcieux A, Francina A, Vital-Durand D, Abnormal hemoglobins with high oxygen affinity in the differential diagnosis of polycythemia]Rev Med Interne 2011 32(10):e105-07.10.1016/j.revmed.2010.11.00621511372 [Google Scholar] [CrossRef] [PubMed]
[13]. Kalotychou V, Tzanetea R, Konstantopoulos K, Papassotiriou I, Rombos I, Erythrocytosis due to a combination of the high oxygen affinity hemoglobin variant, Hb Olympia{β20(B2)Val→Met} with β- and α-thalassemia mutations: First case in the literatureHemoglobin 2010 34(4):383-88.10.3109/03630269.2010.48633120642336 [Google Scholar] [CrossRef] [PubMed]
[14]. Djulbegovic M, Lee AI, Chen K, Which patients with unprovoked venous thromboembolism should receive extended anticoagulation with direct oral anticoagulants? A systematic review, network meta-analysis, and decision analysisJ Eval Clin Pract 2020 26(1):7-17.10.1111/jep.1319431190408 [Google Scholar] [CrossRef] [PubMed]
[15]. Nygaard M, Petersen J, Bjerrum OW, Haemoglobinopathia Ypsilanti- A rare, but important differential diagnosis to polycythaemia veraLeuk Res Rep 2013 2(2):86-88.10.1016/j.lrr.2013.09.00224371790 [Google Scholar] [CrossRef] [PubMed]