Noonan Syndrome (NS) is an autosomal dominant condition caused by mutations in multiple genes in the RAS-MAP (Mitogen-Activated Protein) Kinase pathway. It is typically characterised by short stature, broad webbed neck, abnormal chest shape, congenital heart defects and developmental delay. Oral manifestations include high arched palate, micrognathia, malocclusion, impacted teeth and jaw bones. Presented here is a case of NS in a 26-year-old female, reported to the dental clinic in College of Dentistry, Princess Nourah Bint Abdulrahman University. The cranio-dentofacial features of this syndrome can be diagnosed by the dentist and these features can be unrecognised by the physician. The dentofacial features of the patient included a broad forehead, down slanting palpebral fissure, flat base of the nose and low junction of the ears, prominent nasolabial folds, Class III molar malocclusion, edge-to-edge bite, high arched palate, and congenitally missing teeth. The present patient was first diagnosed by a dental professional and hence, this case report aims to present this syndrome from a dental viewpoint. The treatment plan was to improve her oral hygiene, retain the deciduous tooth and space maintainer in the congenitally missing tooth to preserve space for the future prosthodontic treatment.
Case Report
A 26-year-old Saudi female, born after a normal pregnancy from healthy parents with no family history of genetic disease, came for a regular dental checkup in the dental clinic for females. Her medical history revealed a congenital heart defect, ovarian cyst, eye problems, and she is under regular review in a Governmental Hospital. Her dental history revealed that she had received multiple restorations. Physical examination revealed abnormal chest shape (pectus excavatum), broad webbed neck, lower posterior hair line, with normal nails and fingers and normal stature. Facial features revealed a normal vertical facial pattern, straight facial profile, broad forehead, down slanting palpebral fissure, flat base of the nose and low junction of the ears, prominent nasolabial folds [Table/Fig-1]. Intraoral examination and radiographic examination which was based on (Orthopantomography, Bilateral Bitewing and Selective Periapical Radiographs) revealed she is in the mixed dentition, retained primary tooth (#75), multiple congenitally missing permanent teeth (upper lateral incisors, lower second premolars, left lower second molar, and upper third molars) [Table/Fig-2,3 and 4]. There were a total of 25 permanent teeth present, in the upper dental arch, first and second molars #17, 16, 26, 27, premolars #15, 14, 24, 25, canines #13, 23 and central incisors #11, 21. In the lower dental arch-molars #38, 36, 46, 47, 48, first premolars #34, 44, all the lower anterior teeth #33, 32, 31, 41, 42, 43 and retained primary tooth #75. All present teeth were caries free, fair oral hygiene, Class III incisor malocclusion, edge-to-edge bite with normal sized jaw, high arched palate. Echocardiography report shows myxomatous mitral valve, bileaflet prolapse, anterior mitral valve prolapse of both anterior (A2) leaflets causing moderate Mitral Regurgitation (MR), trace pulmonic valve regurgitation. The left ventricle is normal in size, and Ejection Fraction was 60% which was noted from her medical records. And her eye examination showed Amblyopia, loss of foveal reflex with mild choroid atrophy.
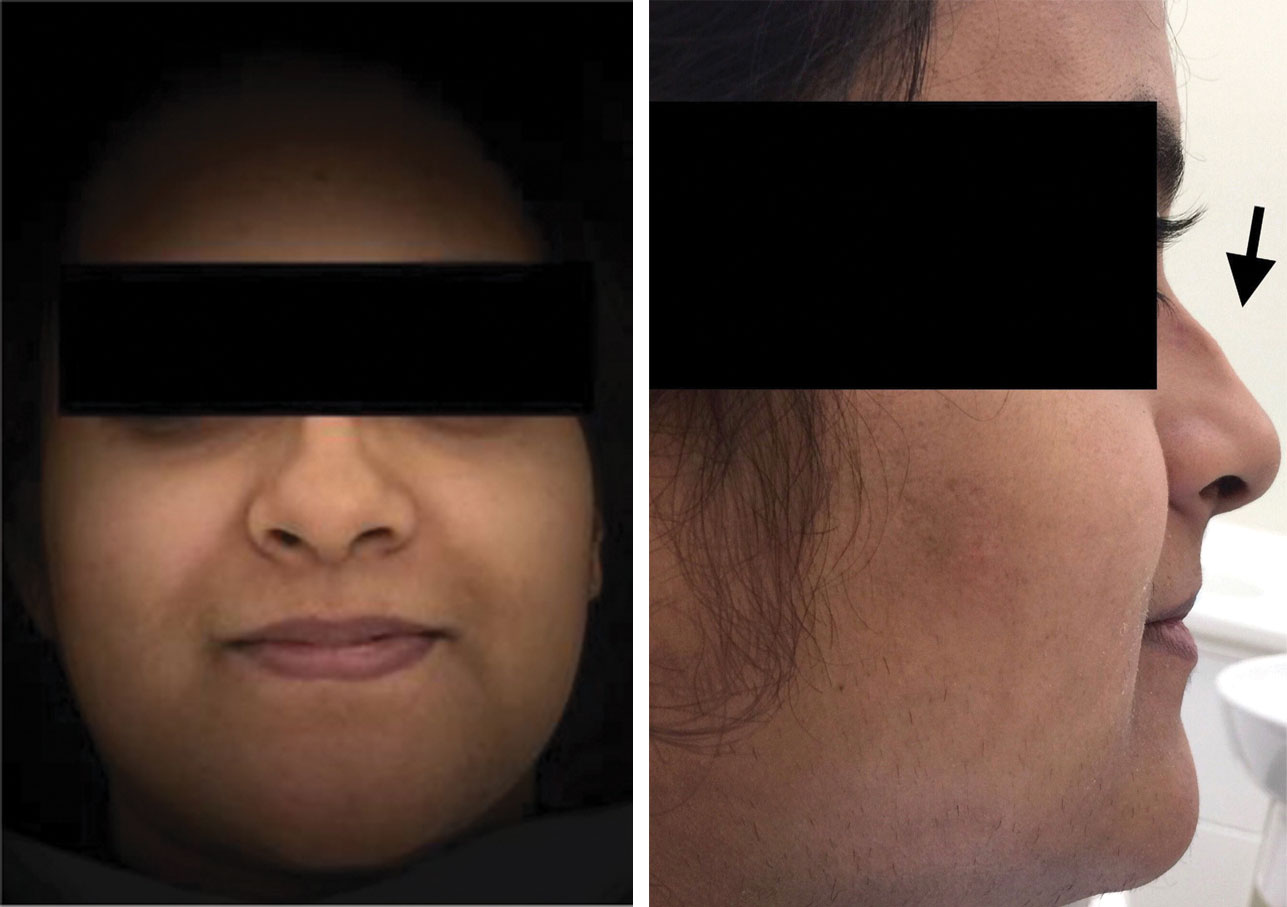
Extra-Oral photograph (frontal view, lateral view) Showing flat nasal bridge, saddle nose, prominent nasolabial folds.
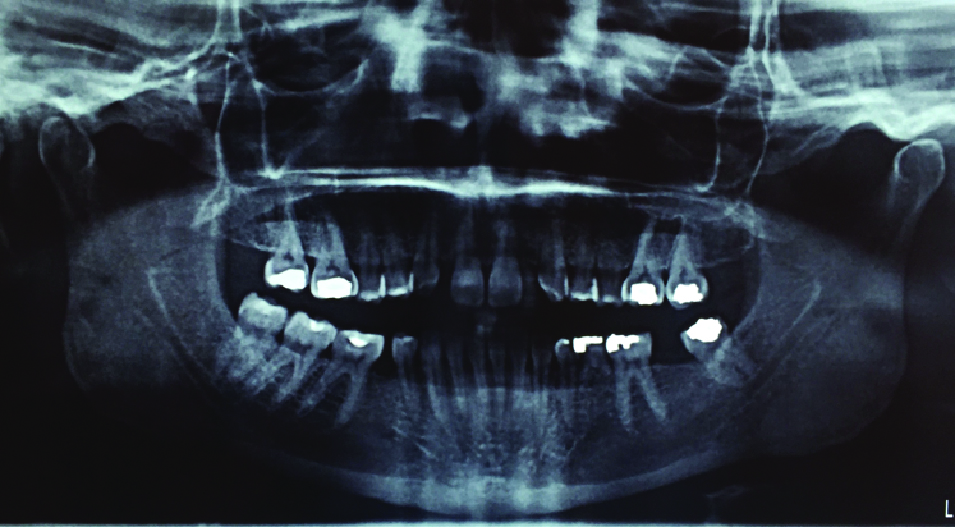
Orthopantogram (OPG) showing congenitally missing teeth (18, 12, 22, 28, 37, 35, 45), no impacted or supernumerary teeth.
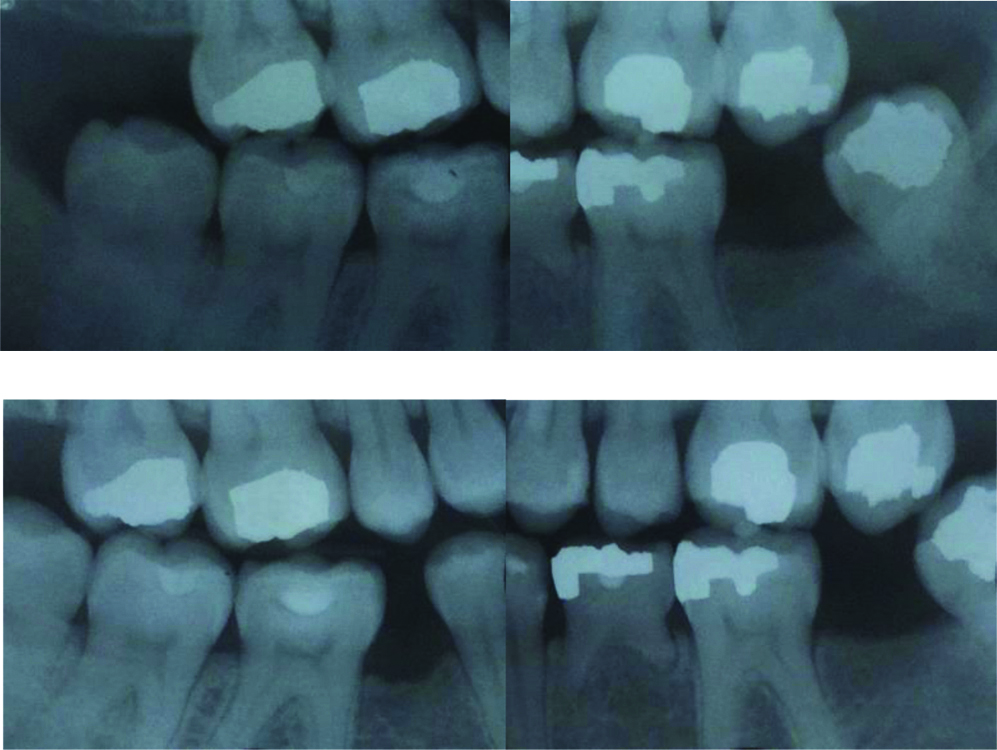
Radiographs depicting lower left primary molar showing no signs of ankyloses and no evidence of caries.
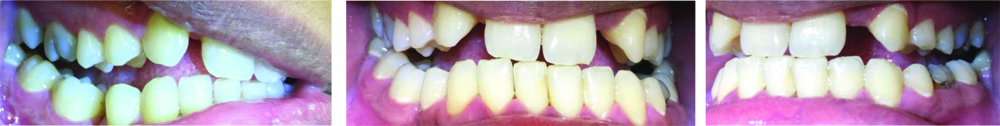
Intra-oral photographs showing retained deciduous tooth #75 and unerupted permanent teeth (18, 12, 22, 28, 37, 35, 45).
The CT/CBCT images were not indicated in this case. Lateral cephalometric radiograph was suggested for orthodontic treatment to aid in diagnosis and treatment planning. Since the patient refused to undergo orthodontic treatment, lateral cephalometric radiograph was not taken. There are several disorders with significant phenotypic overlaps with NS; such as Turner syndrome and Aarskog syndrome [1]. The present case could not be attributed to Aarskog syndrome due to lack of digital and genital findings [2]. The differential diagnosis include Turner Syndrome, because Turner syndrome shares many features with NS [1]. Turner syndrome only affects females (45X karyotype), and hence NS is also known as “Pseudo-Turner syndrome” in females and “Male Turner syndrome” in males [1]. However, due to normal karyotype which is the cardinal feature of this syndrome excludes it from the diagnosis [1]. Diagnosis will be based mainly on clinical features, since mutation testing will prove as a diagnostic in approximately 70% of cases due to the other gene(s) responsible for NS, which are still unknown [3,4]. In this study, the patients refused to do genetic test, so diagnosis was based on clinical features and Van der Burgt diagnostic criteria. According to Van der Burgt criteria in order to make a definitive diagnosis, there should be one major and two minor signs or one minor and two major signs [4-6]. In this case, there were two major (typical facial dysmorphology and chest deformity) and one minor signs (cardiac problems). There are a number of treatment modalities like orthodontic and prosthodontic treatment but patient cooperation is essential before starting any comprehensive dental treatment.
We had to tailor a treatment plan for unmotivated patient with underlying systemic disease who refused to do orthodontic and prosthodontic treatment, and with dental aesthetic satisfaction. Management strategies include maintenance of the existing dentition and facilitation of oral function. In this case, the focus was on her concern which is to preserve her primary tooth. Treatment involved oral prophylaxis, placement of pre-fabricated Stainless Steel Crown (SSC) to aid in the preservation of deciduous tooth #75 as long as possible which requires only one visit and band and loop space maintainer in the congenitally missing left lower second molar area to preserve space for the future prosthodontic treatment, in addition to a proper oral hygiene.
Discussion
The NS is a multisystem disorder occuring in approximately 1 in 1,000 to 2,500 people [7]. The characteristic findings of this syndrome include short stature, chest deformity, hypertelorism, ptosis, congenital heart disease and distinctive facial features such as down slanting palpebral fissure, broad forehead, low junction of the ears and posteriorly rotated [7]. Cohorts studies showed that NS affects male more than female patients [7]. The syndrome caused by mutations in multiple genes in the RAS-MAP Kinase pathway, missense mutations in the PTPN11 gene on chromosome 12 account for upto 50% of cases [7,8]. Recently, mutations in the SOS1, RAF1, RIT1, MEK1, KRAS and NRAS, BRAF, MAP2K1, RRAS, RASA2, A2ML1, SOS2, LZTR1, ADNP, p.Gly503Arg and p.Met504Val genes have been identified in a small proportion of patients with NS [5,7,9,10]. Several studies have demonstrated that this hyperactivation of the phosphatase (gain-of-function mutation) is responsible for heart malformation, growth retardation and craniofacial defects observed in NS [2,11,12].
In 15-30% of the cases, responsible genes and/or mutations have not yet been identified [7]. The oral manifestations of this syndrome include narrow, high arched palate, open bite, malocclusion, greater risk of caries process, late eruption, multiple submerged and retained deciduous teeth, impacted teeth, above average need for orthodontic treatment, articulation difficulties, retroclined mandibular incisors and giant cells in the jaws [1,2,13]. Some of NS cases develop mandibular cysts, which can mimic cherubism [10]. Noonan Syndrome (NS) causes heterogeneous group of multiple congenital anomalies, which are easy to recognise clinically [7]. Even though molecular testing can be performed, the diagnosis of NS can be made on the basis of the clinical picture, based on Van der Burgt diagnostic criteria [1]. Van der Burgt developed diagnostic criteria based on family history, height, as well as features such as facial, cardiac, chest wall and other anomalies [1]. Furthermore, it has been suggested to include oral manifestations into the scoring criteria for the diagnosis of NS [1]. Facial analysis technology can help clinicians in making accurate NS diagnosis based on an elaborated combination of deep surface models from three-dimensional scans combined with pattern recognition algorithms [10]. Facial analysis technology can be a useful complement to the physician’s dysmorphology examination, easier to use and more accessible especially in countries without access to genetic services or genetic testing [10]. To date, there is still no effective specific therapy for NS and other RASopathies, only symptomatic treatments exist for this syndrome [11]. Therefore, patients affected by NS must be followed-up by a multidisciplinary team and a life-long follow-up for late complications are recommended, in order to prevent problems that could become more complicated and untreatable [1,7,11]. The discovery of the crucial role of RAS/MAPK activation in the pathophysiology of NS and other RASopathies, therapeutic strategies aiming to reduce this activation or targeting transcription factors seems to be very promising [2]. Moreover, 3-hydroxy-3-methylglutaryl CoA reductase inhibitors, commonly known as statins, have been suggested as a potential therapy for RASopathies by reducing its localisation to the plasma membrane [11]. Until now, therapeutic approaches still under development and several are currently undergoing clinical trials [11]. Regarding prognosis, most adults with NS do not require special medical care [5]. New medical problems are not expected to appear in adulthood [5]. Some patients may have health problems as a consequence of their congenital heart defect, or other anomaly associated with NS, so regular follow-up with the physician is essential [3,5]. There is no evidence child bearing complication in females with NS [5]. However, genetic counselling is essential because there is upto 50% chance of transmitting it to her children [3]. Before the child is born, parental consultation is essential to address the recurrence risk in the family and the management options [5]. Oral health care for NS patients is necessary starting from the first year of age, in order to prevent problems that could become irreversible and difficult to manage [1]. Moreover, full mouth rehabilitation is inevitable when there is a lack of timely intervention. Full mouth rehabilitation in such patients improves aesthetics, function and comfort, but may require extensive costly treatment to achieve appropriate results. Patients unwilling to come for their dental appointments should be early detected and managed accordingly. Retention of primary molars due to absence of permanent successor cases should be approached with special caution, and multiple factors should be considered. The solution to retain the deciduous tooth and preserve the space could lead to a less complicated dental treatment options in the future and better overall prognosis. The other treatment option was to extract the deciduous tooth and implant placement and space closure of the lower arch spaces using orthodontic treatment. Treatment options for congenitally missing lateral incisors include either space opening with prosthetic replacement or space closure with canine substitution. Finally, the dentist should clarify to the patient and document it in the patients’s record that further treatment is needed and patient should consent in refusal of recommended treatment form.
Conclusion(s)
Patients with NS often exhibit cranio-dentofacial characteristics. Increased awareness among health care providers will lead to correct earlier diagnosis and provide patients and their families with optimal management and counselling. Therefore, they must be followed-up by a multidisciplinary team including dental care, in order to prevent problems that could become more complicated and untreatable.
[1]. Mallineni SK, Yiu CKY, King NM, Oral manifestations of Noonan syndrome: Review of the literature and a report of four casesRom J Morphol Embryol 2014 55(4):1503-09. [Google Scholar]
[2]. Madhuri V, Gopal A, Vinay C, Chandrasekhar R, Uloopi K, Multiple unerupted permanent teeth associated with Noonan syndromeAnn Med Health Sci Res 2015 5(4):31710.4103/2141-9248.16019026229724 [Google Scholar] [CrossRef] [PubMed]
[3]. Emral ME, Akcam MO, Noonan syndrome: A case reportJ Oral Sci 2009 51(2):301-06.10.2334/josnusd.51.30119550102 [Google Scholar] [CrossRef] [PubMed]
[4]. Bhambhani V, Muenke M, Noonan syndromeAm Fam Physician 2014 89(1):37-43. [Google Scholar]
[5]. van der Burgt I, Noonan syndromeOrphanet J Rare Dis 2007 2(4):1750-1172.10.1186/1750-1172-2-417222357 [Google Scholar] [CrossRef] [PubMed]
[6]. Croonen EA, Nillesen W, Schrander C, Jongmans M, Scheffer H, Noordam C, Noonan syndrome: comparing mutation-positive with mutation-negative dutch patientsMol Syndromol 2013 4(5):227-34.10.1159/00035068623885229 [Google Scholar] [CrossRef] [PubMed]
[7]. Tafazoli A, Eshraghi P, Koleti ZK, Abbaszadegan M, Noonan syndrome- A new surveyArch Med Sci 2017 13(1):215-22.10.5114/aoms.2017.6472028144274 [Google Scholar] [CrossRef] [PubMed]
[8]. Siegfried A, Cances C, Denuelle M, Loukh N, Tauber M, Cavé H, Noonan syndrome, PTPN11 mutations, and brain tumors. A clinical report and review of the literatureAm J Med Genet A 2017 173(4):1061-65.10.1002/ajmg.a.3810828328117 [Google Scholar] [CrossRef] [PubMed]
[9]. Carrasco P, Gómez G, Carreto P, Granell R, Vázquez I, León A, Noonan syndrome: Severe phenotype and PTPN11 mutationsMed Clin (Barc) 2019 152(2):62-64.10.1016/j.medcli.2018.03.01529703613 [Google Scholar] [CrossRef] [PubMed]
[10]. Kruszka P, Porras AR, Addissie YA, Moresco A, Medrano S, Mok GTK, Noonan syndrome in diverse populationsAm J Med Genet A 2017 173(9):2323-34.10.1002/ajmg.a.3836228748642 [Google Scholar] [CrossRef] [PubMed]
[11]. Yart A, Edouard T, Noonan syndrome: An update on growth and developmentCurr Opin Endocrinol Diabetes Obes 2018 25(1):67-73.10.1097/MED.000000000000038029120925 [Google Scholar] [CrossRef] [PubMed]
[12]. Poaty H, Mbika-Cardorelle A, Moukouma C, Mouko A, Clinical diagnosis of noonan syndrome and brief review of literatureAnn Med Health Sci Res 2017 7:76-79. [Google Scholar]
[13]. Cao H, Alrejaye N, Klein OD, Goodwin AF, Oberoi S, A review of craniofacial and dental findings of the RASopathiesOrthod Craniofac Res 2017 20(1):32-38.10.1111/ocr.1214428643916 [Google Scholar] [CrossRef] [PubMed]