Intra-abdominal Solitary Myofibroma in a Child: A Rare Case Report
Smita Singh1, Jyoti Garg2, Kusha Sharma3, Kiran Agarwal4
1 Professor, Department of the Pathology, Lady Hardinge Medical College, New Delhi, India.
2 Assistant Professor, Department of the Pathology, Lady Hardinge Medical College, New Delhi, India.
3 Senior Resident, Department of the Pathology, Lady Hardinge Medical College, New Delhi, India.
4 Director Professor, Department of the Pathology, Lady Hardinge Medical College, New Delhi, India.
NAME, ADDRESS, E-MAIL ID OF THE CORRESPONDING AUTHOR: Kusha Sharma, C-2/2001, Vasant Kunj, New Delhi-110070, India.
E-mail: drkushasharma@gmail.com
Intra-abdominal masses in children are usually malignant. Benign tumours at this location are not seen frequently. It is even rarer to find solitary myofibromas intra-abdominally as these tumours are known to have predilection for the head and neck region. We present an unusual case of solitary myofibroma with abdominal localisation in a six-year-old male child who presented with abdominal distension since six months. Computed Tomography (CT) abdomen revealed a complex solid-cystic mass extending from umbilicus to right iliac fossa and measuring 9×6×4 cm. Histopathological examination revealed a tumour displaying biphasic nodular pattern with the presence of lighter staining fascicles of mature myoid cells along with darker staining and more cellular areas of smaller primitive cells. On Immunohistochemistry (IHC), tumour cells were positive for vimentin and Smooth Muscle Actin (SMA) with variable reactivity for desmin while these were negative for CD34 and Anaplastic Lymphoma Kinase 1 (ALK). Based on the histopathological and immunohistochemical findings, final diagnosis of myofibroma was made. Recognition of these lesions is extremely challenging owing to their rare presentation intra-abdominally and also because of their close morphological overlap with other spindle cell tumours commonly found at this site. This case highlights the combined role played by histopathology and IHC in making a clear distinction between different entities. It is imperative for both clinicians and histopathologists to establish the correct diagnosis as excision of the solitary myofibroma is curative in most cases and offers better clinical course than the more commonly found malignant tumours at this site.
Childhood, Myofibromatosis, Myopericytoma
Case Report
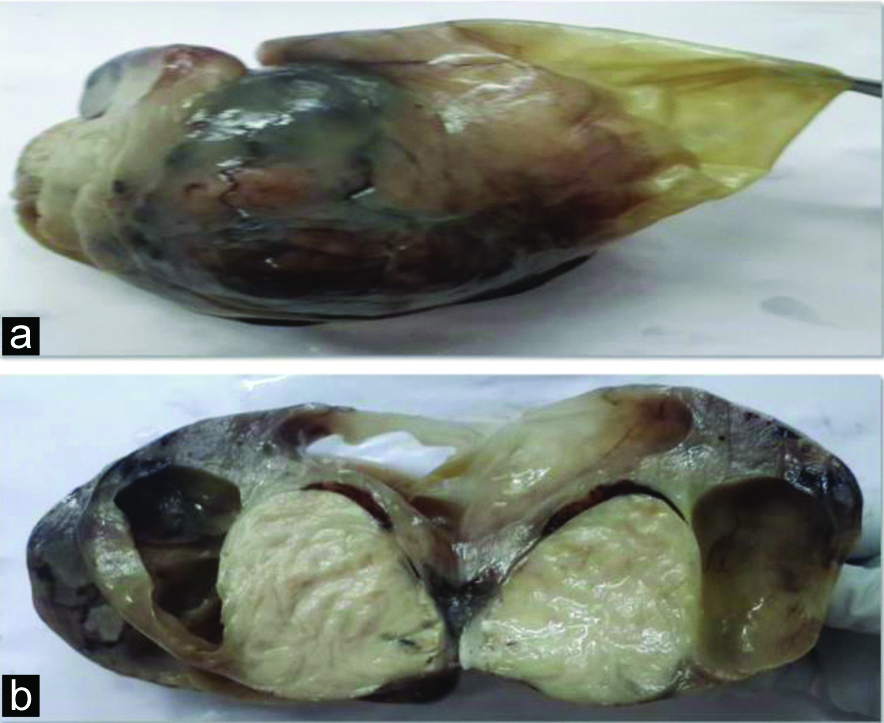
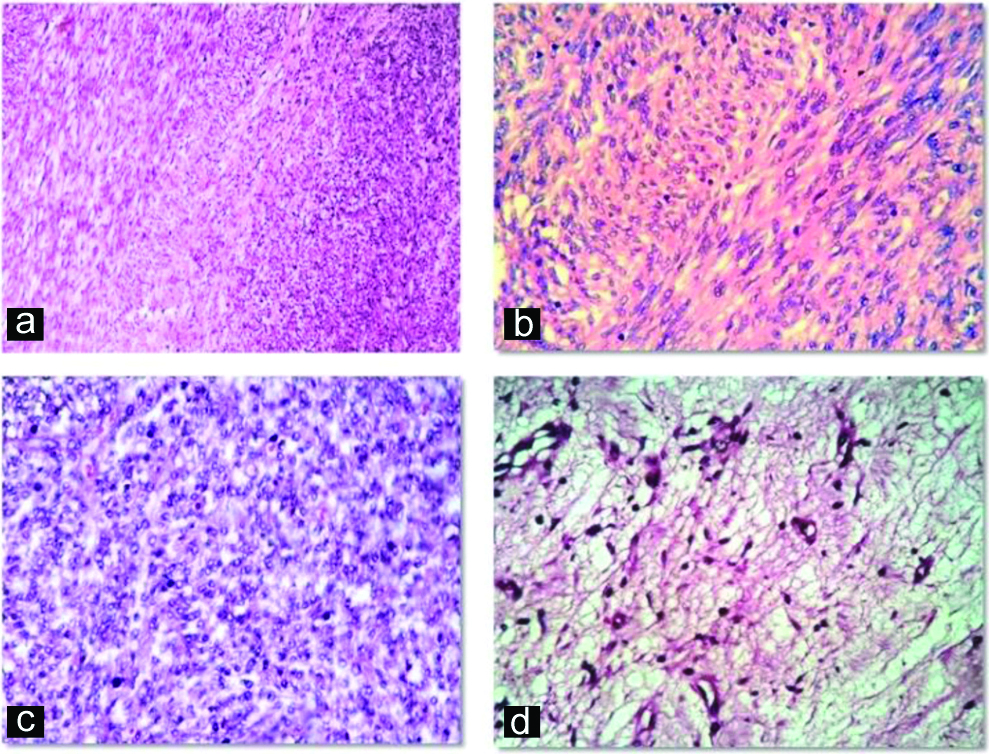
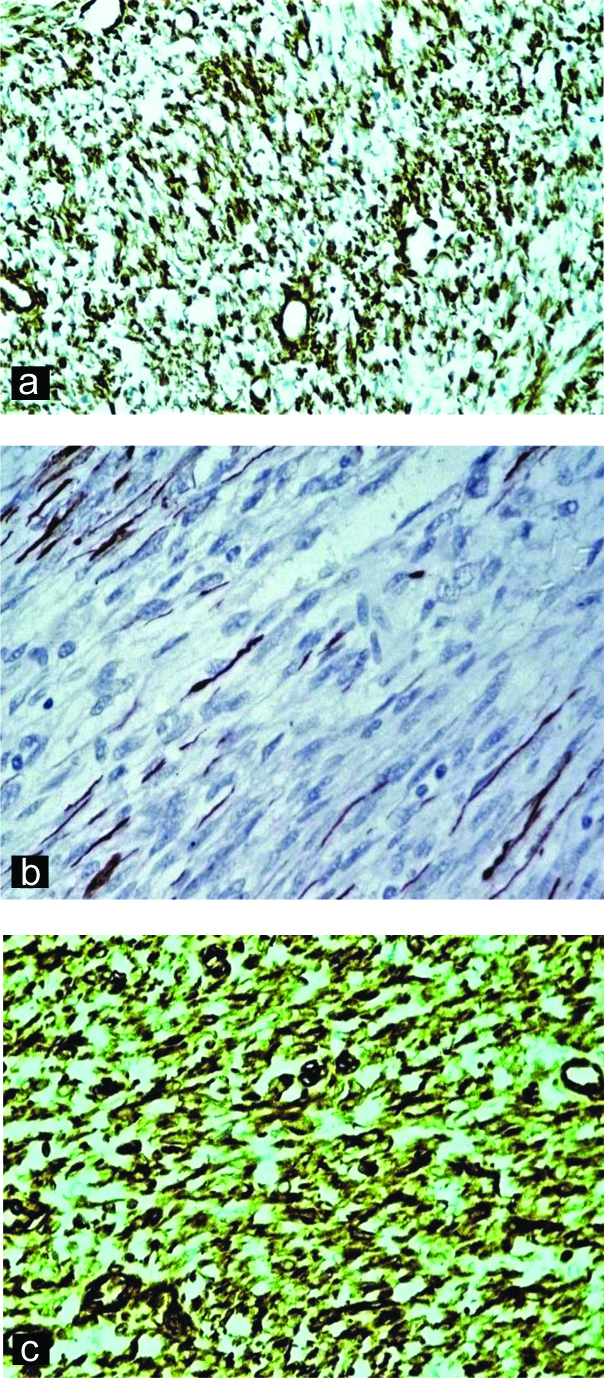
A six-year-old male child presented with complaints of abdominal distension for six months duration with no other significant history. Abdominal examination revealed an ill-defined mass in the right iliac fossa. CT abdomen showed a complex solid-cystic mass extending from umbilicus to right iliac fossa and measuring 9x6x4 cm. Provisional diagnosis of mesenchymal tumour with differential diagnosis of inflammatory myofibroblastic tumour, solitary fibrous tumour and leiomyoma were considered. Preoperatively, mass was found to be intraperitoneal with no firm adhesions to any viscera and was enucleated easily. Grossly, the mass was well-circumscribed [Table/Fig-1a] with solid-cystic cut surface [Table/Fig-1b]. Cysts were 4-5 in number, measured 0.5-4 cm and contained serous fluid. Solid areas were grey-white and firm. Histopathological examination showed a well-circumscribed tumour with a pseudo-capsule. Tumour showed a biphasic nodular pattern with the presence of lighter staining fascicles of mature myoid cells along with darker staining and more cellular areas of smaller primitive cells [Table/Fig-2a]. Myoid cells had moderate amount of eosinophilic cytoplasm and elongated, tapering nuclei with vesicular chromatin [Table/Fig-2b]. Smaller cells had ovoid, dark nuclei and scant indistinct cytoplasm [Table/Fig2c]. At places, these cells were arranged around hemangiopericytoma-like vessles. Areas of cystic degeneration, myxoid change [Table/Fig-2d] were seen. No cytological atypia or increased mitotic activity were noted. Various spindle cell tumours were considered as differentials including myofibroma, leiomyoma, inflammatory myofibroblastic tumour and solitary fibrous tumour. On IHC, tumour cells exhibited positivity for SMA [Table/Fig-3a], variable reactivity for desmin [Table/Fig-3b] and positivity for vimentin [Table/Fig-3c]. Tumour cells were negative for CD34 and ALK. A final diagnosis of myofibroma was thus made. Since the lesion was solitary, simple excision was curative. Patient showed no recurrence at one year follow-up.
a) Gross specimen showing a well-circumscribed mass (9x6x4 cm); b) Cut surface showing both solid and cystic areas.
a) Tumour cells arranged in biphasic nodular pattern with paler staining myoid cells along with darker staining immature cells (H&E, 100X); b) Fascicles of spindle cells with eosinophilic cytoplasm, elongated tapering vesicular nuclei (H&E, 400X); c) Nodule of smaller cells with round to ovoid nuclei and scant cytoplasm (H&E, 400X); d) Myxoid change (H&E, 100X).
a) Tumour cells positive for Smooth Muscle Actin (SMA) (SMA, 400X); b) Tumour cells showing variable reactivity for Desmin (Desmin, 400X); c) Tumour cells positive for Vimentin (Vimentin, 400X).
Discussion
Intra-abdominal tumours in children are usually malignant with neuroblastoma being the commonest amongst these and having incidence of 10.2 cases per million children under 15 years of age [1]. Benign intra-abdominal tumours are less common and can pose a diagnostic challenge. Myofibroma is a benign tumour composed of myofibroblastic cells, is a subtype of myopericytoma and belongs to the family of pericytic/perivascular tumour [2]. It can occur as a solitary form or multicentric form (myofibromatosis) [3]. Solitary myofibromas usually present as a cutaneous or a subcutaneous mass of the head and neck region [4,5]. It has been very rarely reported intra-abdominally. Infantile myofibromatosis, a mesenchymal disorder of early childhood, is characterised by the formation of myoid cells with thin walled blood vessels in the skin, muscle, viscera, bone, and subcutaneous tissue [6]. Solitary lesions usually have a benign clinical course while the multicentric form sometimes involves bone and viscera and the mortality rate approaches 70% with visceral involvement [7]. It is therefore important to correctly diagnose and effectively treat such cases as they usually have good prognosis.
Myofibromatosis as an entity was originally described as “congenital generalised fibromatosis” in 1954 by Stout [7]. The term “infantile myofibromatosis”, was introduced by Chung EB and Enzinger FM, to distinguish it from more aggressive types of fibromatosis, reflecting the young age of onset of this disease and the myofibroblastic nature of the tumour cells [2]. Daimaru Y et al., coined the term “myofibromatosis” [8]. Finally, these terms were adopted by the World Health Organisation (WHO), 2002 to describe the solitary form (myofibroma) and multicentric forms (myofibromatosis) [5]. In the current edition of WHO within the pericytic (perivascular) tumours, hemangiopericytoma is abolished as presence of hemangiopericytoma like vessels is just not limited to pericytic tumours but also described in other lesions unrelated to this category [9,10].
Most of these tumours present in the first two years of life. The disease is more common in males. Approximately, half of solitary myofibromas occur in the cutaneous or subcutaneous tissue of the head and neck region followed by trunk and upper and lower extremities. The remaining half occur in deep-seated structures, such as skeletal muscle, aponeuroses, and bone. Myofibromatosis involves both soft tissue and bone and in upto 15 to 20% of cases deep soft tissue and visceral lesions are seen. Visceral involvement is rare and found in less than 4% of the solitary lesions [5]. In the present case, the patient was a six-year-old male and the lesion was solitary, intraperitoneal in location and did not involve any visceral organ.
Grossly, lesions vary in size from 0.5 cm to 7 cm (median- 2.5 cm). On cut surface, myofibromas are firm, fibrous, grey-white in colour, often having necrotic areas and/or cysts filled with caseous material or haemorrhage [5]. In this case, the lesion was well-circumscribed, measuring 9x6x4 cm and had grey-white, firm, solid cut surface along with small cystic areas filled with serous fluid.
At low magnification, they show a biphasic growth pattern, consisting of fascicles or whorls of myoid-appearing spindle cells and vascular spaces with a hemangiopericytoma-like appearance [11]. The myoid-appearing spindle cells have eosinophilic cytoplasm and elongated, tapering nuclei with a vesicular chromatin and 1-2 small nucleoli. The primitive cells are round or polygonal with relatively scant cytoplasm and are arranged around thin-walled, irregularly branching, hemangiopericytoma-like blood vessels. There is no significant atypia or pleomorphism and mitotic activity is minimal. Immunohistochemical findings support the myofibroblastic origin of the tumour as they are positive for vimentin and SMA with a variable reactivity for desmin [3,5]. Although desmin reactivity is usually negative in myofibromas, it has been described. In a study by Oudijk L et al., 7/72 tumours were positive for desmin and showed significant association with size of lesion, the median size of lesion with and without Desmin reactivity were 42 mm and 21 mm, respectively [12]. This may correlate with findings of the present study as the lesion was 9×6×4 cm and showed variable staining for desmin.
The differential diagnoses of myofibroma includes various types of mesenchymal tumours, including leiomyoma, nodular fascitis, inflammatory myofibroblastic tumour, solitary fibrous tumour, infantile fibrosarcoma and paediatric leiomyosarcoma. Intra-abdominal leiomyoma occurs exclusively in females and are rarely reported in children. They lack the typical biphasic pattern and hemangiopericytoma-like vessels seen in myofibroma and are diffusely positive for SMA and desmin. Nodular fasciitis is characterised by a mucin-rich stroma rendering most lesions a ‘tissue culture-like’ or ‘feathery’ appearance and frequently shows increased mitotic activity, inflammatory infiltrate and giant cells [13]. It lacks biphasic pattern and hemangiopericytoma-like vessels. All these features were not seen in the present case. Inflammatory myofibroblastic tumour was ruled out due to lack of inflammatory infiltrate and ALK negativity. Solitary fibrous tumour shows hemangiopericytoma-like pattern of the vascular network, which is similar to that seen in myofibroma [14]. However, it was not considered in this case due to biphasic pattern of the tumour, presence of smaller primitive cells and negativity for CD34. Infantile fibrosarcoma is infiltrative, arranged in intersecting fascicles usually exhibiting a herring-bone pattern with tumoural cells showing minimal pleomorphism and prominent mitotic activity. The lesion in the present case was non-infiltrative, showed a biphasic pattern and did not show increased or atypical mitoses thereby ruling out infantile fibrosarcoma. Recent literature reveals a small subset of myofibromas displaying atypical features like hypercellularity, poorly formed myoid nodules, infiltrative growth pattern and perineural invasion which may confuse with fibrosarcoma [15]. Further confirmation can be made as most infantile fibrosarcomas possess a chromosomal translocation causing ETV6-NTRK3gene fusion transcript which is not associated with myofibroma. Paediatric leiomyosarcomas typically show blunt ended cigar shaped nuclei arranged in long fascicles intersecting at right angles and exhibit more cellular pleomorphism and higher mitotic rate along with diffuse positivity for desmin. This was convincingly excluded in this case.
Recently, recurrent somatic Platelet-Derived Growth Factor Receptor-Beta (PDGFRB) mutations have been reported in a large number of myofibroma cases [16]. A novel subset of cellular variants of myofibromas/myopericytomas harbouring SRF-RELA fusions and showing smooth muscle-like immunophenotype have also been described [17]. These tumours displayed worrisome histologic features, such as increased cellularity, solid or focally infiltrative growth, increased mitotic activity (>10/10 HPF) in few cases and strong and diffuse co-expression of SMA and desmin in most cases.
Although myofibroma and myofibromatosis are defined as a benign fibroblastic/myofibroblastic tumours, the biological behaviour of these lesions is determined by the pattern of organ involvement and not by histologic features. However, solitary or multiple lesions confined to soft tissues and bone have an excellent prognosis. The local recurrence rates are reported to be less than 10% for solitary lesions. The lesions tend to undergo spontaneous regression or be cured by simple local excision [3].
Conclusion(s)
Solitary myofibroma should be kept in the differential diagnoses of spindle cell tumours in paediatric age group even in locations other than head and neck. Worrisome histological features such as hypercellularity and necrosis can lead to a misdiagnosis, if this entity is not kept in mind.
Author Declaration:
Financial or Other Competing Interests: None
Was informed consent obtained from the subjects involved in the study? Yes
For any images presented appropriate consent has been obtained from the subjects. Yes
Plagiarism Checking Methods: [Jain H et al.]
Plagiarism X-checker: Sep 09, 2020
Manual Googling: Nov 20, 2020
iThenticate Software: Dec 21, 2020 (9%)
[1]. Maris JM, Recent advances in neuroblastomaN Engl J Med 2010 362(23):2202-11.10.1056/NEJMra080457720558371 [Google Scholar] [CrossRef] [PubMed]
[2]. Mentzel T, Bridge JA, Myopericytoma, including myofibroma. In: Fletcher CD, Hogendoorn CW, Mertens F, Bridges J, edsWHO Classification of Tumours of Soft Tissue and Bone 2013 4th edLyon, FranceIARC Press:118-20. [Google Scholar]
[3]. Chung EB, Enzinger FM, Infantile myofibromatosisCancer 1981 48(8):1807-18.10.1002/1097-0142(19811015)48:8<1807::AID-CNCR2820480818>3.0.CO;2-G [Google Scholar] [CrossRef]
[4]. Choi JH, Ro JY, Cutaneous spindle cell neoplasms pattern-based diagnostic approachArch Pathol Lab Med 2018 142(8):958-72.10.5858/arpa.2018-0112-RA30040457 [Google Scholar] [CrossRef] [PubMed]
[5]. Fletcher CDM, Unni KK, Mertens F, World health organisation classifictionof tumoursPathology and Genetics of Tumours of Soft Tissue and Bone 2002 LyonIARC Press [Google Scholar]
[6]. Hicks J, Mierau G, The Spectrum of Pediatric Fibroblastic and MyofibroblasticTumoursUltrastructural Pathology 2004 28(5-6):265-81.10.1080/01913129088210515764576 [Google Scholar] [CrossRef] [PubMed]
[7]. Stout AP, Juvenile fibromatosesCancer 1954 7(5):953-78.10.1002/1097-0142(195409)7:5<953::AID-CNCR2820070520>3.0.CO;2-W [Google Scholar] [CrossRef]
[8]. Daimaru Y, Hashimoto H, Enjoji M, Myofibromatosis in adults (adult counterpart of infantile myofibromatosis)Am J Surg Pathol 1989 13(10):859-65.10.1097/00000478-198910000-000052782546 [Google Scholar] [CrossRef] [PubMed]
[9]. WHO Classification of Tumours Editorial BoardSoft tissue and bone tumours 2020 Lyon (France)International Agency for Research on Cancer(WHO classification of tumours series, 5th ed. Vol. 3) [Google Scholar]
[10]. Sbaraglia M, Bellan E, Dei Tos AP, The 2020 WHO Classification of Soft Tissue Tumours: news and perspectivesPathologicaEpub 2020 Nov 3. https://doi.org/10.32074/1591-951X-21310.32074/1591-951X-213 [Google Scholar] [CrossRef]
[11]. Flucke U, Karanian M, Broek RWT, Thway K, Soft tissue special issue: perivascular and vascular tumours of the head and neckHead Neck Pathol 2020 14:21-32.https://doi.org/10.1007/s12105-020-01129-z10.1007/s12105-020-01129-z31950476 [Google Scholar] [CrossRef] [PubMed]
[12]. Oudijk L, den Bakker MA, Hop WCJ, Cohen M, Charles A, Alaggio R, Solitary, multifocal and generalised myofibromas: Clinicopathological and immunohistochemical features of 114 casesHistopathology 2012 60(6B):01-11.10.1111/j.1365-2559.2012.04221.x22486319 [Google Scholar] [CrossRef] [PubMed]
[13]. Dayan D, Nasrallah W, Vered M, Clinico-pathologic correlations of myofibroblastic tumours of the oral cavityJ Oral Pathol Med 2005 34(7):426-35.10.1111/j.1600-0714.2005.00338.x16011613 [Google Scholar] [CrossRef] [PubMed]
[14]. Guillou L, Fletcher JA, Fletcher CD, Mandahl N, Extrapleural solitary fibrous tumour and hemangiopericytoma. In: Fletcher CD, Unni KK, Mertens F, edsWHO classification of tumours. Pathology and genetics. Tumours of soft tissue and bone 2002 LyonIARC Press:86-88. [Google Scholar]
[15]. Linos K, Carter JM, Gardner JM, Folpe AL, Weiss SW, Edgar MA, Myofibromas with atypical features: expanding the morphologic spectrum of a benign entityAm J Surg Pathol 2014 38:1649-54.10.1097/PAS.000000000000027024921644 [Google Scholar] [CrossRef] [PubMed]
[16]. Agaimy A, Bieg M, Michal M, Geddert H, Märkl B, Seitz J, Recurrent somatic PDGFRB mutations in sporadic infantile/solitary adult myofibromas but not in angioleiomyomas and myopericytomasAm J Surg Pathol 2017 41(2):195-203.10.1097/PAS.000000000000075227776010 [Google Scholar] [CrossRef] [PubMed]
[17]. Antonescu CR, Sung YS, Zhang L, Agaram NP, Fletcher CD, Recurrent SRF-RELA fusions define a novel subset of cellular myofibroma/ myopericytoma: A potential diagnostic pitfall with sarcomas with myogenic differentiationAm J Surg Pathol 2017 41(5):677-84.10.1097/PAS.000000000000081128248815 [Google Scholar] [CrossRef] [PubMed]